F.-Y. Dupradeau, C. Cezard, R. Lelong, E. Stanislawiak, J. Pecher, J. C. Delepine & P. Cieplak, R.E.DD.B.: A database for RESP and ESP atomic charges, and force field libraries, Nucl. Acids Res. (Database issue), 2008, 36, D360-D367, [Abstract], [Local PDF file].
This tutorial describes the RESP ESP charge DDataBase, or R.E.DD.B., and its different tools. Submission procedures to R.E.DD.B. involving whole molecule(s) and molecule fragment(s) are first presented. The submission of projects obtained from the previous tutorial are taken as examples. The submission of more complex projects, which are already available in R.E.DD.B. are then briefly commented. Finally, the update and the download of R.E.DD.B. projects are described.
 Description of R.E.DD.B.
Description of R.E.DD.B. 
-I.1. Description of a R.E.DD.B. project
-I.2. Submission of projects to R.E.DD.B.
-I.2.1- Submission of "molecule fragment" projects
-I.2.1.1- Submission of a "molecule fragment" project involving a single molecule
-I.2.1.2- Submission of a "molecule fragment" project involving two molecules
-I.2.1.3- Examples of more complex projects
-I.2.2- Submission of "whole molecule" projects
-I.2.2.1- Submission of a "whole molecule" project involving a single molecule
-I.2.2.2- Submission of a "whole molecule" project involving several different molecules
-I.3.- Update of projects in R.E.DD.B.
-I.4.- List and download of R.E.DD.B. projects
-I.4.1- List of R.E.DD.B. projects
-I.4.2- Download of R.E.DD.B. projects
Description of the R.E.DD.B. database
Please, first read the R.E.DD.B. Frequently Asked Questions, which provide complementary information about R.E.DD.B. and its different tools.
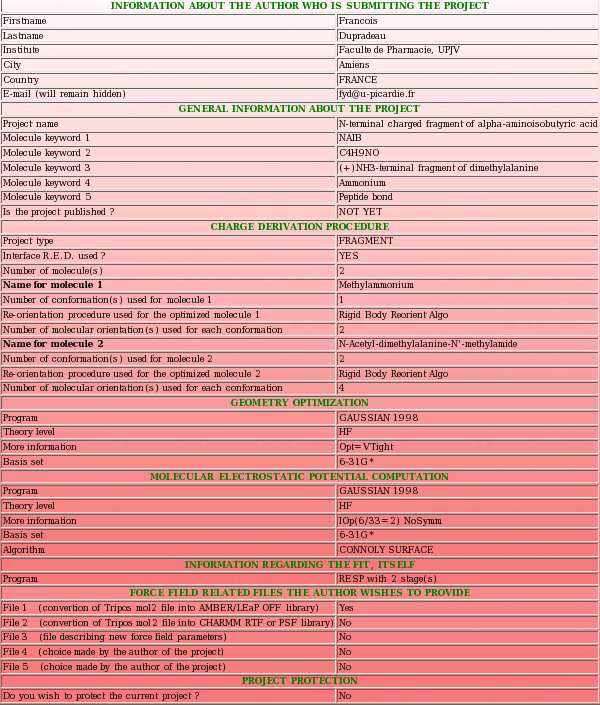
The RESP ESP charge DDataBase, or R.E.DD.B., is a unique source of molecular electrostatic potential based (RESP and ESP) atomic charge values embedded in force field libraries for small molecules and molecule fragments. R.E.DD.B. allows storing and distributing RESP and ESP charge values in the scientific community. These charges are of high quality and reproducibility (see FAQ-2). However, R.E.DD.B. not only stores RESP and ESP atomic charge values, but also the structures and information about the procedure used to derive the reported charges. Force field parameters can also be provided by the author along with the force field libraries. These pieces of information are gathered in R.E.DD.B. projects (see FAQ-1), and each R.E.DD.B. project is identified by a R.E.DD.B. code (see FAQ-3) ensuring an efficient system of classification.
A R.E.DD.B. project ensures the reproducibility of the reported charge values and the rigorous description of the corresponding atomic charge derivation procedure. Indeed, RESP or ESP charge derivation involving multiple molecules, multiple conformations and/or multiple orientations is an error-prone and complex task. Thus, in our opinion, storing the information about the computational conditions of such approaches is as important as the charge values themself.
Before anything else, one has to well understand the concept of a R.E.DD.B. project, and which information/files can be found in a R.E.DD.B. project.
-I.1- Description of a R.E.DD.B. project
Two types of project named "whole molecule" and "molecule fragment" projects (see FAQ-1 for more information) can be found in R.E.DD.B. A R.E.DD.B. project is referenced by a R.E.DD.B. code attributed after that the project has been officially recorded in R.E.DD.B. Whole molecule and molecule fragment R.E.DD.B. codes are constituted by the "W-" and "F-" letters followed by a number respectively representing the total number of whole molecule and molecule fragment projects available in R.E.DD.B. at the time the new project is recorded in R.E.DD.B. (see FAQ-3).
A R.E.DD.B. project contains (i) the atomic charge values for the whole molecule(s) or molecule fragment(s) studied, (ii) the Cartesian coordinates of the corresponding whole molecule(s), and (iii) the computational conditions used in the charge derivation procedure:
(i) The atomic charge values and the topology of a whole molecule or molecule fragment are provided in the Tripos mol2 file format (see FAQ-7; § -7.2- b) and FAQ-9 for more information). A single Tripos mol2 file is required to fully characterize a whole molecule or a molecule fragment, independently of the number of conformation(s) and molecular orientation(s) used in the charge derivation.
(ii) By opposition to the Tripos mol2 file format, a PDB file contains information about the number of conformation(s) and molecular orientation(s) of a whole molecule used in the charge derivation. In fact, the Cartesian coordinates optimized by quantum mechanics (QM) of the different conformations representing a whole molecule are provided to the PDB format in a single file (see FAQ-7; § -7.2- a) and FAQ-9).
(iii) The information about the computational conditions used in the charge derivation are available in a "Summary of information" (see FAQ-7; § -7.1- and FAQ-9). Input(s) for the program used to fit the atomic charge values to the molecular electrostatic potential(s) are also available (see FAQ-7; § -7.2- c) and FAQ-9).
(iv) Optional files describing force field related information might also be available in a R.E.DD.B. project (see FAQ-7; § -7.3- and FAQ-9).
-I.2- Submission of projects to R.E.DD.B.
A project submitted to R.E.DD.B. is now always involved in a "peer review" system: researchers working in the field of computational chemistry have agreed to participate in the evaluation of R.E.DD.B. projects. After project submission and reviewing by two referees the author has to correct her/his abstract/data in agreement with the referee comments. Thus this is only after reviewing and data correction that a project is made available in R.E.DD.B. (see FAQ-5).
Scripts, which help to prepare project submission to R.E.DD.B. are now available.
First, one has to "Register" from the R.E.DD.B. main page as a R.E.DD.B. user to be able to submit/upload a project to R.E.DD.B. After registration, a new R.E.DD.B. user is going to automatically receive an email with her/his R.E.DD.B. 'login' and 'password'. Then, just click on the "Submit project" R.E.DD.B. tool available from the R.E.DD.B. main page to start a project submission to R.E.DD.B.
However, before to start a submission to R.E.DD.B. it is advised to:
- Read the FAQ-5 where the submission procedure is described,
- Study projects already available in R.E.DD.B.,
- Prepare the files and information required for the project one wishes to submit to R.E.DD.B. before to start the submission (See FAQ-7), and
- Perform blind tests in the R.E.DD.B.2t (Bis & test) Internet site.
- In case of problem, please, ask your questions in the AMBER or CCL mailing list, using for instance this type of title/header: "R.E.DD.B. query: Your question..." (see FAQ-6).
After clicking on the "Submit project" R.E.DD.B. tool, a new page entitled "SUBMIT A NEW PROJECT" is opened (see Picture 1). By clicking on the two "Warning" panels, new popup windows show up providing general information about the R.E.DD.B. Internet site (see Picture 2) and about setting the preferences in your favorite browser (see Picture 3 and FAQ-11).
 |
 |  | |
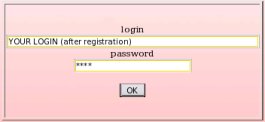
Then, just provide the R.E.DD.B. login and password received by email after registration, and click on the "OK" button (see Picture 4).
 |
-I.2.1- Submission of "molecule fragment" projects
Let us directly start by submitting "molecule fragment" projects to R.E.DD.B. This will allow us characterizing the differences between a "whole molecule" and a "molecule fragment" R.E.DD.B. project.
-I.2.1.1- Submission of a "molecule fragment" project involving a single molecule
Different examples of RESP charge derivation leading to a molecule fragment and involving a single molecule were described in the section "-I.2.2.3-" of the previous tutorial. They correspond to the R.E.D. III job number (No.) "-13-" to "-16-", and to the "F-1", "F-2" and "F-3" R.E.DD.B. projects. In this new tutorial, the project submission to R.E.DD.B. which corresponds to the R.E.D. III job No. "-16-" and to the "F-3-" R.E.DD.B. project is going to be described as a first example. In the job No. "-16-", two-conformations (alpha helix and extended) for the Dimethylalanine dipeptide molecule and four-molecular orientations for each conformation were involved in RESP charge derivation, and two intra-molecular charge constraints were used during the fit to lead to the definition of the central fragment of Dimethylalanine (see "Scheme 1" from the previous tutorial).
After providing a login and password, a new page entitled "SUBMIT A NEW PROJECT 1/4" is automatically opened (see Picture 5).
 |
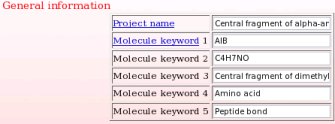
One starts the project submission by providing general information about the current project (see Picture 6).
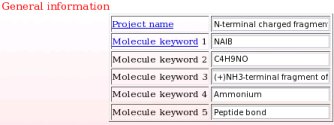
- This includes a project name and up to five project keywords. A description for each piece of information required during a project submission is available through popup windows (see Picture 7 describing the help one can read about the project name for instance). The project name is used in the creation of a link to the Wikipedia encyclopedia (see FAQ-9; § -9.6-), and the project keywords allow the search of projects in R.E.DD.B. using the "Download projects" R.E.DD.B. tool (see FAQ-10). In the present example (R.E.D. III job No. "-16-"), the project name chosen is "Central fragment of alpha-aminoisobutyric acid", and the keywords selected are "AIB", "C4H7NO", "Central fragment of dimethylalanine", "Amino acid" and "Peptide bond".
 |  | |
- A summary describing the project has to be written (see Picture 8). This summary contains a detailed description of the charge derivation procedure used to calculate the charge values reported.
 |
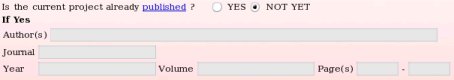
- The reference of the publication describing the R.E.DD.B. project or any work which uses the R.E.DD.B. project can be provided if known (see Picture 9). However, it might be more suitable to submit a project to R.E.DD.B. before its publication. Thus, the corresponding project code(s) generated by R.E.DD.B. can be incorporated in the manuscript before it is sent to the journal editor. Such approach allows the establishment of a rigorous connection between the publication reference and the R.E.DD.B. code(s) (see FAQ-3 for more information). In these conditions, the publication reference can be provided after the project submission in R.E.DD.B. using the "Update project" R.E.DD.B. tool available from the R.E.DD.B. main page (see FAQ-12 for more information). In the present example, the publication reference is not provided since still unknown.
 |
- A R.E.DD.B. project can be protected for a period of 3, 6 or 12 months (see Picture 10) meaning that only the author and the R.E.DD.B. superuser can have access to the protected data during the period of protection (see FAQ-4 for more information). By default, a R.E.DD.B. project is not protected. In the present example, we decided to not protect the "F-3" project allowing every R.E.DD.B. user to study this project.
 |
- If the author wishes, she/he can also provide force field related information compatible with the R.E.DD.B. project submitted (see Picture 11). This can be scripts allowing the convertion of Tripos mol2 file(s) into AMBER OFF or CHARMM RTF libraries, new force field parameters or any information the author decides to provide (see FAQ-7; § -7.3- for more information). As R.E.DD.B. is mainly devoted to RESP and ESP charge values, the default is to not provide such force field related files. In the present example, only a LEaP script allowing the convertion of the Tripos mol2 file provided into an AMBER OFF force field library is going to be uploaded.
 |
The next step corresponds to the description of the charge derivation procedure, itself.
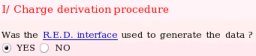
- One starts by answering to the question if the R.E.D. interface was used to generate the R.E.DD.B. project (see Picture 12). Projects which were not executed by R.E.D. can obviously be submitted to R.E.DD.B. However, the files [PDB, Tripos mol2 and fitting program input(s)] required for a R.E.DD.B. submission are automatically generated by the R.E.D. program. Thus, the use of R.E.D. makes a project submission in R.E.DD.B. straightforward. In the present example, the R.E.D. program version III was used.
 |
- One has to select the type of R.E.DD.B. project submitted (see Picture 13), i. e. is it a "whole molecule" or a "molecule fragment" project (see FAQ-1 for more information) ? In the present example, the project submitted is a "molecule fragment" project. Indeed, the charge derivation procedure applied leads to the derivation of RESP charge values for the central fragment of Dimethylalanine starting from the Dimethylalanine dipeptide molecule.
 |
- One has to provide the number of (whole) molecule(s) used in the charge derivation procedure (see Picture 14; first question). For each whole molecule, (i) a name, (ii) the number of conformation(s), (iii) the re-orientation procedure applied before the molecule electrostatic potential (MEP) computation and (iv) the number of molecular orientation(s) used in MEP computation is going to be requested. For each whole molecule, a PDB file specific of the molecule has to be uploaded. Besides the four pieces of information just reported, a PDB file also contains the Cartesian coordinates optimized by QM (i. e. generated during a geometry optimization) for the different conformations of a whole molecule (see FAQ-7; § -7.2- a)). In the present example, a single whole molecule (represented by two conformations and four molecular orientations) was used in the charge derivation procedure. Consequently, a single PDB file automatically generated by R.E.D. III (named "File4REDDB_m1.pdb", see TABLE 1) has to be uploaded to fully describe the Dimethylalanine dipeptide molecule.
One has also to provide the number of Tripos mol2 file(s) one wishes to upload (see Picture 14; second question). This/these file(s) contain the charge values and describe the topology of the target "whole molecule(s)" or "molecule fragment(s)" generated during the charge derivation procedure. The Tripos mol2 file format does not contain any information about the conformation(s) and molecule orientation(s) used in the charge derivation procedure. Since the whole molecule name and the molecule fragment name differ, a name has also to be provided for the structure represented by a Tripos mol2 file (see FAQ-7; § -7.2- b)). In the present example, the type of R.E.DD.B. project is a "molecule fragment" project. Consequently, a Tripos mol2 file representing this central molecule fragment has to be uploaded. Although four Tripos mol2 files were generated for the job No. "-16-", (i. e. one by conformation for the Dimethylalanine dipeptide molecule, and one by conformation for the central fragment of Dimethylalanine), a single set of RESP charge values (compatible with the two conformations and four orientations used in the charge derivation procedure) were generated for the target fragment. Consequently, a single Tripos mol2 file has to be uploaded (named "Mol_m1-o1-sm.mol2" or "Mol_m1-o2-sm.mol2"; see TABLE 1) to fully describe the charge values and the topology of the fragment studied.
 |
When ready, one clicks on the "Next" button to go to the next upload page, or on the "Cancel" button to go back to the previous upload page (see Picture 15).
 |
After clicking on the "Next" button, a new page entitled "SUBMIT A NEW PROJECT 2/4" is automatically opened (see Picture 16).
 |
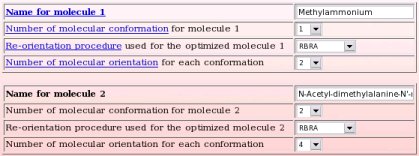
- We start this new upload page by describing the whole molecule(s) (which is/are going to be provided to the PDB format; see TABLE 1) used in the charge derivation procedure. As just said, for each whole molecule, (i) a name (international nomenclature; a link to the Wikipedia encyclopedia is created for each whole molecule name), (ii) the number of conformation(s), (iii) the re-orientation procedure applied before the MEP computation and (iv) the number of molecular orientation(s) used in MEP computation have to be provided. In the present example, we have previously answered that a single whole molecule was used in the charge derivation procedure. Consequently, a single set of information is required (see Picture 17): The molecule name chosen is "N-Acetyl-dimethylalanine-N'-methylamide", the number of conformations used in the charge derivation procedure is equal to two, the molecular re-orientation procedure used before the MEP computation is based on the Rigid-body re-orientation algorithm (RBRA) implemented in the R.E.D. program, and the number of molecular orientations used in the MEP computation is equal to four.
 |
Then, the QM calculations used in the charge derivation procedure have to be described (see Pictures 18 and 19).
- The computational conditions used in the first step of the charge derivation procedure, i. e. the geometry optimization step of the different conformations of the different whole molecules used in the charge derivation procedure are provided (see Picture 18). In the present example, the "Gaussian 1998" program, the "HF/6-31G*" theory level and the particular "Opt=VTight" keyword were used.
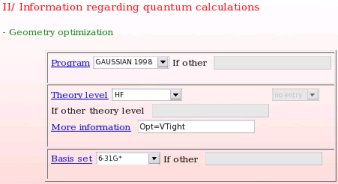 |
- The computational conditions used in the second step of the charge derivation procedure, i. e. the MEP computation step of the different molecular orientations of the different conformations used in the charge derivation procedure are provided (see Picture 19). In the present example, the "Gaussian 1998" program, the "HF/6-31G*" theory level, the Connolly surface algorithm, and the particular "IOp(6/33=2) NoSymm" keywords were used.
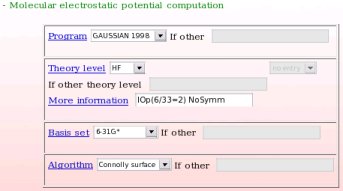 |
Then, the fitting conditions used in the thrid and last step of the charge derivation have to be described (see Picture 20). In the present example, the "RESP program" was used to fit the charge values to the MEP, and the RESP fit was performed in two stages. Since a two stage RESP fit was used, the two corresponding inputs are going to be requested to accuratly describe the fitting step (see FAQ-7; § -7.2- for more information).
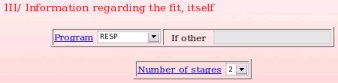 |
When ready, one clicks on the "Next" button to go to the next upload page, or on the "Previous" button to go back to the previous upload page and to perform some checking and/or corrections (see Picture 21).
 |
After clicking on the "Next" button, a new page entitled "SUBMIT A NEW PROJECT 3/4" is automatically opened (see Picture 22).
 |
- Then, a summary of the information provided so far is displayed (see Picture 23). It has to be carefully checked by its author before the upload of the different files corresponding to the current project in R.E.DD.B. If the user realizes that she/he made an error, she/he can use the "Previous" button (see Picture 29) to go back to the previous upload page and to correct her/his mistakes(s).
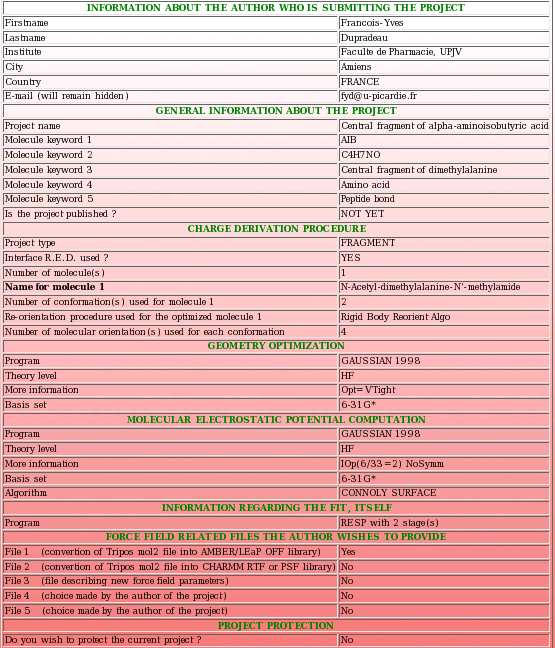 |
After double checking, one is ready to upload the files corresponding to the information just provided (see Picture 24).
 |
Let us list the PDB (whole molecule), Tripos (whole molecule and molecule fragment) and RESP input files generated by R.E.D. III for the job No. "-16-" (two-conformation four-orientation RESP fit). Among those files, only the File4REDDB_m1.pdb, Mol_m1-o1-sm.mol2 (or Mol_m1-o2-sm.mol2), input1_m1.sm and input2_m1.sm files are required to fully describe the current R.E.DD.B. project:
| Job No. | RESP charge derivation | |||
| -16- | Two-conf. four-orient | File4REDDB_m1.pdb Mol_m1-o1-qmra.pdb Mol_m1-o1-rbra1.pdb Mol_m1-o1-rbra2.pdb Mol_m1-o1-rbra3.pdb Mol_m1-o1-rbra4.pdb Mol_m2-o1-qmra.pdb Mol_m2-o1-rbra1.pdb Mol_m2-o1-rbra2.pdb Mol_m2-o1-rbra3.pdb Mol_m2-o1-rbra4.pdb |
Whole molecule Mol_m1-o1.mol2 Mol_m1-o2.mol2 Molecule fragment Mol_m1-o1-sm.mol2 Mol_m1-o2-sm.mol2 |
Without intra-molecular charge constraint input1_m1 input2_m1 With intra-molecular charge constraints input1_m1.sm input2_m1.sm |
- Tripos mol2 files are first uploaded (see Picture 25). One has previously declared that a single Tripos mol2 file corresponding to the central fragment of Dimethylalanine would be uploaded. Since the name of this fragment was not provided yet, it is provided at this stage of the project submission procedure. Finding an IUPAC name for a molecule fragment might not be as usual as for a whole molecule. Consequently, more common name for a molecule fragment might be chosen such as "Central fragment of alpha-aminoisobutyric acid". As for each whole molecule name, for each molecule fragment name, a link to the Wikipedia encyclopedia is automatically created. The upload of the Tripos mol2 file is performed by browsing the file system tree and selecting the file "Mol_m1-o1-sm.mol2" or "Mol_m1-o2-sm.mol2" (see TABLE 1) generated by the R.E.D. III program.
 |
- PDB files are then uploaded (see Picture 26). One has previously declared that a single PDB file corresponding to the Dimethylalanine dipeptide molecule would be uploaded. The name of this whole molecule being already known, it is printed in bold just in front of the button allowing the upload of the corresponding file. The upload of the PDB file is performed by browsing the file system tree and selecting the file "File4REDDB_m1.pdb" (see TABLE1) generated by the R.E.D. III program.
 |
- The input(s) of the fitting program used in the third step of the charge derivation are then uploaded (see Picture 27). One has previously declared that a two RESP stage was performed. The upload of the two RESP input files is performed by browsing the file system tree and selecting the "input1_m1.sm" and "input2_m1.sm" files.
 |
- The force field related file(s) compatible with the project being submitted that the author wishes to provide are finally uploaded (see Picture 28). One has previously declared that only a LEaP script allowing the convertion of the Tripos mol2 file provided into an AMBER OFF force field library would be uploaded. The corresponding script file which is manually written is uploaded (script1.ff).
 |
When ready the user clicks on the "Upload" button to upload the five files required (see Picture 29), or on the "Previous" button to upload the previous page and to correct the error(s) she/he made.
 |
After clicking on the "Upload" button, a last page entitled "SUBMIT A NEW PROJECT 4/4" is automatically opened (see Picture 30).
 |
- A summary of the upload process is printed (see Picture 31). When all the files are successfully uploaded the "[Done]" word is printed for each file. If a single file is not successfully uploaded (because of an error in the file system path for instance), the "[FAILED]" word is printed, and nothing is recorded in R.E.DD.B. Consequently, one has to correct the project by using the "Return to the previous page to complete this project" button, or restart the project submission all over from the beginning.
 |
- A message explaining that the project submitted in R.E.DD.B. was successfully uploaded is then printed (see Picture 32).
 |
- A short summary describing the R.E.DD.B. project is printed, and the R.E.DD.B. code is provided (see Picture 33). An email that summarizes these pieces of information is then automatically sent to the author of the project.
 |
 |
CONCLUSION
This tutorial example demonstrated the project submission of a single molecule fragment generated from a single whole molecule. The corresponding project is already recorded in R.E.DD.B. as the "F-3" R.E.DD.B. code. The "Summary of information" corresponding to this project can be obtained by clicking from "here". The data corresponding to these project can be also downloaded using the "Download projects" R.E.DD.B. tool available from the R.E.DD.B. main page.
-I.2.1.2- Submission of a "molecule fragment" project involving two molecules
Different examples of RESP charge derivation leading to a molecule fragment and involving two molecules were described in the section "-I.2.2.4-" of the previous tutorial. They correspond to the R.E.D. III job No. "-18-" and "-19-", and to the "F-7" and "F-11" R.E.DD.B. projects. The project submission which corresponds to the R.E.D. III job No. "-18-" and to the "F-7-" R.E.DD.B. project is going to be described as a new example in this tutorial. The R.E.D. III job No. "-18-" corresponds to the derivation of RESP charge values for the (+)NH3-terminal fragment of Dimethylalanine using two molecules (i. e. "Methylammonium", and "Dimethylalanine dipeptide"). This is achieved using an inter-molecular charge constraint (sets to a value of zero) between the CH3 group of atoms of Methylammonium and the CH3CO-NH group of atoms of Dimethylalanine dipeptide, and using an intra-molecular charge constraint (sets to a value of zero) for the NHCH3 group of atoms (NME residue) of Dimethylalanine dipeptide (see "Scheme 2" from the previous tutorial).
Only the differences between the previous (corresponding the "F-3" R.E.DD.B. code) and the current (corresponding the "F-7" R.E.DD.B. code) examples of this tutorial are now going to be discussed.
After providing a login and password, a new page entitled "SUBMIT A NEW PROJECT 1/4" is opened (see Picture 5). As in the previous example of this tutorial, one has first to provide general information about the project submitted (see Picture 35 compared to Picture 6 from the previous example). A new project name and new keywords compatible with the current project are provided.
 | | |
- A new summary describing the current project has to be written (see Picture 36 compared to Picture 8).
 | | |
- The major difference between the former and the current examples of project submission in R.E.DD.B. is the number of (whole) molecules used in the charge derivation procedure corresponding to the R.E.D. III job No. "-16-" and "-18-". Indeed, these two projects correspond to a "molecule fragment" project because they both leads to the charge derivation for a molecule fragment. However, in the former example, a single molecule was used in the charge derivation while in the current one two molecules were used (see Picture 37 compared to Picture 14 from the previous example). Consequently, one has to take into account of this difference when one submits the current project to R.E.DD.B.
 | | |
After clicking on the "Next" button, a new page entitled "SUBMIT A NEW PROJECT 2/4" is opened (see Picture 16).
- In the present example (R.E.D. III job No. "-18-"; "F-7" R.E.DD.B. project), two (whole) molecules were used in the charge derivation procedure. Consequently, two sets of information are required (see Picture 38 compared to Picture 17 from the previous example).
For the first molecule, the name chosen is "Methylammonium", the number of conformations used in the charge derivation procedure is equal to one, the molecular re-orientation procedure used before the MEP computation is based on the Rigid-body re-orientation algorithm (RBRA) implemented in the R.E.D. program, and the number of molecular orientations used in the MEP computation is equal to two.
For the second molecule, the name chosen is "N-Acetyl-dimethylalanine-N'-methylamide", the number of conformations used in the charge derivation procedure is equal to two, the molecular re-orientation procedure used before the MEP computation is based on the Rigid-body re-orientation algorithm (RBRA) implemented in the R.E.D. program, and the number of molecular orientations used in the MEP computation is equal to four.
 | | |
- The QM and the fitting conditions used in the charge derivation procedure for the R.E.D. III job No. "-16-" and "-18-" are identical (see Pictures 18, 19 and 20).
After clicking on the "Next" button, a new page entitled "SUBMIT A NEW PROJECT 3/4" is opened (see Picture 22). Then, a summary of the information provided so far is displayed (see Picture 39).
 |
After double checking, one is ready to upload the files corresponding to the information provided (see Picture 24).
- A single Tripos mol2 file representing the (+)NH3-terminal fragment of Dymethylalanine generated during the charge derivation has to be upload. The name chosen for this molecule fragment is "N-terminal charged fragment of alpha-aminoisobutyric acid" (see Picture 40).
Let us list the Tripos mol2 files generated by R.E.D. III for the job "-18-", and the single file required for the current project submission to R.E.DD.B. (among those files only the "Mol_m2-o1-mm1.mol2" or "Mol_m2-o2-mm1.mol2" file needs to be uploaded):
[fyd@lynx 6-Charges-2conf-RBRA-4-INTER-MCC1]$ ls */*.mol2
| Mol_m1/Mol_m1-o1.mol2 | Whole molecule No. 1 |
| Mol_m2/Mol_m2-o1.mol2 | Whole molecule No. 2 / Conf. No. 1 |
| Mol_m2/Mol_m2-o1-sm.mol2 | Molecule 2 fragment (use intra-molecular charge constraints) / Conf. No. 1 |
| Mol_m2/Mol_m2-o2.mol2 | Whole molecule No. 2 / Conf. No. 2 |
| Mol_m2/Mol_m2-o2-sm.mol2 | Molecule 2 fragment (use intra-molecular charge constraints) / Conf. No. 2 |
| Mol_MM/Mol_m2-o1-mm1.mol2 | (+)NH3-terminal fragment of Dimethylalanine (using Conf. 1 of Molecule 2) => R.E.DD.B. |
| Mol_MM/Mol_m2-o2-mm1.mol2 | (+)NH3-terminal fragment of Dimethylalanine (using Conf. 2 of Molecule 2) |
 |
- The two PDB files representing the two whole molecules used the charge derivation, one for "Methylammonium", the other for "Dimethylalanine dipeptide" (see Picture 41) are uploaded.
Let us list the PDB files generated by R.E.D. III for the job "-18-", and the two files required for the current project submission to R.E.DD.B. (among those files only the "File4REDDB_m1.pdb" and "File4REDDB_m2.pdb" files need to be uploaded):
[fyd@lynx 6-Charges-2conf-RBRA-4-INTER-MCC1]$ ls */*.pdb
| Mol_m1/File4REDDB_m1.pdb | Whole molecule No. 1 (single conf., with information about RBRA) => for R.E.DD.B. |
| Mol_m1/Mol_m1-o1-qmra.pdb | Optimized Cart. coordinates from QM output (QMRA) / Molecule 1 |
| Mol_m1/Mol_m1-o1-rbra1.pdb | Optimized Cart. coordinates after RBRA / Molecule 1 (single Conf.), Orient. 1 |
| Mol_m1/Mol_m1-o1-rbra2.pdb | Optimized Cart. coordinates after RBRA / Molecule 1 (single Conf.), Orient. 2 |
| Mol_m2/File4REDDB_m2.pdb | Whole molecule No. 2 (two conf., with information about RBRA) => for R.E.DD.B. |
| Mol_m2/Mol_m2-o1-qmra.pdb | Optimized Cart. coordinates from QM output (QMRA) / Molecule 2, Conf. 1 |
| Mol_m2/Mol_m2-o1-rbra1.pdb | Optimized Cart. coordinates after RBRA / Molecule 2, Conf. 1, Orient. 1 |
| Mol_m2/Mol_m2-o1-rbra2.pdb | Optimized Cart. coordinates after RBRA / Molecule 2, Conf. 1, Orient. 2 |
| Mol_m2/Mol_m2-o1-rbra3.pdb | Optimized Cart. coordinates after RBRA / Molecule 2, Conf. 1, Orient. 3 |
| Mol_m2/Mol_m2-o1-rbra4.pdb | Optimized Cart. coordinates after RBRA / Molecule 2, Conf. 1, Orient. 4 |
| Mol_m2/Mol_m2-o2-qmra.pdb | Optimized Cart. coordinates from QM output (QMRA) / Molecule 2, Conf. 2 |
| Mol_m2/Mol_m2-o2-rbra1.pdb | Optimized Cart. coordinates after RBRA / Molecule 2, Conf. 2, Orient. 1 |
| Mol_m2/Mol_m2-o2-rbra2.pdb | Optimized Cart. coordinates after RBRA / Molecule 2, Conf. 2, Orient. 2 |
| Mol_m2/Mol_m2-o2-rbra3.pdb | Optimized Cart. coordinates after RBRA / Molecule 2, Conf. 2, Orient. 3 |
| Mol_m2/Mol_m2-o2-rbra4.pdb | Optimized Cart. coordinates after RBRA / Molecule 2, Conf. 2, Orient. 4 |
 |
- The two RESP input(s) used in the fitting step of the charge derivation are then uploaded (see Picture 42).
Let us list the RESP input files generated by R.E.D. III for the job "-18-", and the two files required for this project submission to R.E.DD.B. (among those files only the "input1_mm" or "input2_mm" files need to be uploaded):
[fyd@lynx 6-Charges-2conf-RBRA-4-INTER-MCC1]$ ls */input*
| Mol_m1/input1_m1 | Molecule 1: Stage 1. Single molecule, single Conf., two Orient. RESP fit |
| Mol_m1/input2_m1 | Molecule 1: Stage 2. Single molecule, single Conf., two Orient. RESP fit |
| Mol_m2/input1_m2 | Molecule 2: Stage 1. Single molecule, two Conf., four Orient. RESP fit without intra-molecular charge constraint |
| Mol_m2/input1_m2.sm | Molecule 2: Stage 1. Single molecule, two Conf., four Orient. RESP fit with intra-molecular charge constraints |
| Mol_m2/input2_m2 | Molecule 2: Stage 2. Single molecule, two Conf., four Orient. RESP fit without intra-molecular charge constraint |
| Mol_m2/input2_m2.sm | Molecule 2: Stage 2. Single molecule, two Conf., four Orient. RESP fit with intra-molecular charge constraints |
| Mol_MM/input1_mm | Two-molecule RESP fit with intra- & inter-molecular charge constraints: Stage 1 => R.E.DD.B. |
| Mol_MM/input2_mm | Two-molecule RESP fit with intra- & inter-molecular charge constraints: Stage 2 => R.E.DD.B. |
 |
- The LEaP script (script1.ff) allowing the conversion of the Tripos mol2 file into an AMBER OFF force field library is finally uploaded.
 |
After clicking on the "Upload" button, a last Internet page entitled "SUBMIT A NEW PROJECT 4/4" is opened (see Picture 30).
- A summary of the upload process is printed (see Picture 44).
 |
- A short summary describing the R.E.DD.B. project is printed (see Picture 45). The R.E.DD.B. code is provided, and an email is automatically sent to the author to summarize these pieces of information.
 |
CONCLUSION
This tutorial example demonstrated the project submission of a single molecule fragment generated from two whole molecules. The corresponding project is already recorded in R.E.DD.B. as the "F-7" R.E.DD.B. code. The "Summary of information" corresponding to this project can be obtained from "here". The data corresponding to these project can be also downloaded using the "Download projects" R.E.DD.B. tool available from the R.E.DD.B. main page.
-I.2.1.3- Example of more complex projects
More complex projects are available in R.E.DD.B., the "F-45" up to "F-56", and "F-60" R.E.DD.B. projects being perfect examples. A characteristic example is the "F-60" project where nine (whole) molecules were used to lead to thirty two nucleotide fragments. The molecules used in RESP charge derivation are the following:
- Dimethylphosphate (conformation gauche, gauche; 4 mol. orientations),
- the four regular DNA nucleosides (conformations C2'endo and C3'endo; 6 mol. orientations for each conformation), and
- the four regular RNA nucleosides (conformation C3'endo, dihedral constraints were used in the geometry optimization step to avoid intra-molecular hydrogen bonds and transition state type structures: HO3'-O3'-C3'-C4' = 180 deg., HO2'-O2'-C2'-C3' = 290 deg.; 6 mol. orientations).
In such an approach, two inter-molecular charge constraints are used between each methyl group of Dimethylphosphate and the HO5' and HO3' hydroxyl groups of the different nucleosides (see "Scheme 4" from the previous tutorial). Inter-molecular charge equivalencing involving the different deoxyribose and ribose atoms (but the C1' and H1' atoms) is also applied.
Nine PDB files located in "Mol_m$n" sub-directories ("File4REDDB_m$n.pdb" file names; $n = 1 to 9), thirty two Tripos mol2 files [only the "DX" and "RX" central fragments are generated by R.E.D. III, X = A, C, G, T and U (all the fragments are now generated by R.E.D. IV) located in the "Mol_MM" sub-directory, and two multi-molecule multi-conformation multi-orientation RESP inputs also located in the "Mol_MM" sub-directory were uploaded:
- The whole molecule names selected were: Dimethylphosphate, Deoxyadenosine, Deoxycytidine, Deoxyguanosine, Deoxythymidine, Adenosine, Cytidine, Guanosine and Uridine (most of them provide a correct link in the Wikipedia encyclopedia (see FAQ-9; § -9.6-).
- The nucleotide fragment names chosen were: Fragment DA3, Fragment DA5, Fragment DA, Fragment DAN, Fragment DC3, Fragment DC5, Fragment DC, Fragment DCN, Fragment DG3, Fragment DG5, Fragment DG, Fragment DGN, Fragment DT3, Fragment DT5, Fragment DT, Fragment DTN, Fragment RA3, Fragment RA5, Fragment RA, Fragment RAN, Fragment RC3, Fragment RC5, Fragment RC, Fragment RCN, Fragment RG3, Fragment RG5, Fragment RG, Fragment RGN, Fragment RU3, Fragment RU5, Fragment RU, and Fragment RUN (all of them lead to broken links in Wikipedia, but articles might be initiated by the R.E.DD.B. project authors to further develop Wikipedia (see FAQ-9; § -9.6-).
New projects should be soon uploaded in R.E.DD.B. involving new charge derivation and fragment building approaches. When ready this tutorial part will be further developed.
-I.2.2- Submission of "whole molecule" projects
Let us continue this tutorial by submitting "whole molecule" projects to R.E.DD.B.
-I.2.2.1- Submission of a "whole molecule" project involving a single molecule
Different examples of charge derivation involving a single (whole) molecule [represented or not by several conformation(s) and/or molecular orientation(s)] leading to charge values for this molecule were described in the sections "-I.2.2.1-" and "-I.2.2.2-" of the previous tutorial. Examples of such approaches correspond to the R.E.D. III job No. "-1-" to "-11-" (none of the data corresponding to the job No. "-1-" to "-11-" were submitted to R.E.DD.B.). Let us take the job No. "-11-" as a new example for a hypothetical whole molecule project submission to R.E.DD.B. In this example, two conformations (alpha helix and extended) and four molecular orientations for each conformation were used in the charge derivation of the Dimethylalanine dipeptide whole molecule.
After reading the sections -I.2.1.1-, -I.2.1.2-, and -I.2.1.3- of this tutorial, the submission of a whole molecule project in R.E.DD.B. should be straighforward. The major difference, when one wishes to submit a whole molecule project to R.E.DD.B. involving a single whole molecule (compared to a molecule fragment project), is that the tripos mol2 file generated by R.E.D. contains charge values and topology information for a whole molecule (and not anymore for a molecule fragment). Indeed, by definition, the charge derivation procedure corresponding to a whole molecule project involving a single (whole) molecule simply leads to charge values for this molecule. In this case, no molecule fragment originating from the elimination of chemical groups or from the fusion between two molecules is generated. As two project submissions in R.E.DD.B. involving molecular fragments were already detailed and compared (see sections -I.2.1.1-, -I.2.1.2- in this tutorial), only differences with the two former examples are now going to be described:
- After providing general information about the project itself (name, keywords and summary), one has obviously to declare that the project type is, this time, a whole molecule project (see Picture 46 compared to Picture 13 from the two former examples).
 |
- In the present example, the number of whole molecule(s) used in the charge derivation procedure is equal to one (see Picture 14; first question). A name has to be provided for the dimethyalanine dipeptide whole molecule ("N-Acetyl-dimethylalanine-N'-methylamide" could be this name). A single PDB file, "File4REDDB_m1.pdb", generated by the R.E.D. III program for the job No. "-11-" has to be uploaded. As previously described, this file has to contain information about the number of conformation(s) used in the charge derivation procedure with the corresponding Cartesian coordinates optimized by QM, the re-orientation procedure applied before the MEP computation, and the number of molecular orientation(s) for each conformation used in MEP computation.
In the present example, a single Tripos mol2 file, "Mol_m1-o1.mol2" or "Mol_m1-o2.mol2" generated by the R.E.D. III program for the job No. "-11-", has to be uploaded (see Picture 14; second question). This file contains the atomic charge values and describes the topology of the target whole molecule generated during the charge derivation procedure. On the contrary, it does not contain any information about the number of conformation(s) and molecule orientation(s) used in the charge derivation procedure. Finally, since the Tripos mol2 file represent the same structure (i. e. a whole molecule) than the PDB file, the "N-Acetyl-dimethylalanine-N'-methylamide" whole molecule name is used to describe the structure available in both Tripos mol2 and PDB files.
CONCLUSION
This tutorial example quickly demonstrated the project submission to R.E.DD.B. of a single whole molecule. The project corresponding to the job No. "-11-" (from the previous tutorial) is not recorded in R.E.DD.B. However, examples of similar whole molecule projects are available in R.E.DD.B. A good example is the "W-58" R.E.DD.B. project where three conformations (C5, C7ax and C7eq) and ten molecular orientations for each conformation were used in the charge derivation of "N-Acetyl-L-alanine-N'-methylamide". The "Summary of information" corresponding to this project can be obtained from "here". The data corresponding to this project can also be downloaded using the "Download projects" R.E.DD.B. tool available from the R.E.DD.B. main page.
-I.2.2.2- Submission of a "whole molecule" project involving several different molecules
An example of charge derivation involving $n ($n integer > 0) whole molecules [represented or not by several conformation(s) and/or molecular orientation(s)] leading to charge values for these $n whole molecules was described in the job No. "-12-" (section "-I.2.2.2-" from the previous tutorial). The corresponding project was not submitted to R.E.DD.B. However, this example, where $n = 3 whole molecules, might not be the best choice for demonstrating the submission to R.E.DD.B. of a whole molecule project involving several (whole) molecules. Indeed, in this RESP charge derivation example, the different conformations of Dimethylalanine dipeptide are considered as different whole molecules, making this example ambiguous.
Instead, let us describe the submission to R.E.DD.B. of a whole molecule project involving ten different whole molecules, the "W-46" R.E.DD.B. project being a perfect example. In this whole molecule R.E.DD.B. project (see Picture 46), the RESP charge values of ten solvent molecules were derived starting from these ten (whole) molecules. Consequently, one has to declared the number of whole molecules used in the charge derivation procedure and the number of Tripos mol2 file one wishes to upload (see Picture 47).
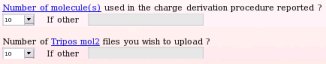 |
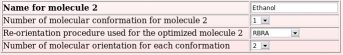
The names of these ten whole molecules have to be provided ("Dimethylsulfoxide", "Ethanol", "Trifluoroethanol", "Methanol", "Acetone", "Acetic acid", "Acetonitrile", "Benzene", "Toluene" and "Chloroform"), with the number of conformation(s) (a single conformation was used whatever the whole molecule choice) and molecule orientation(s) (two or four molecule orientations were generated using the RBRA algorithm) used in the charge derivation for each whole molecule (see Picture 48).
 |
 |
 |
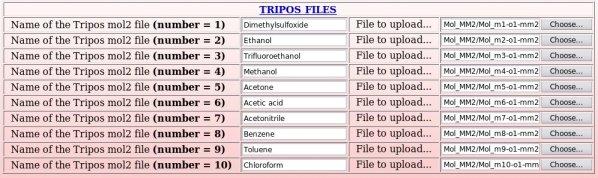
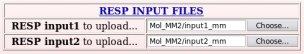
Ten Tripos mol2 files, "Mol_m$n-o1-mm2.mol2", located in the "Mol_MM" sub-directory, ten PDB files, "File4REDDB_m$n.pdb", located in $n "Mol_m$n" sub-directories ($n = 1 to 10) and two RESP inputs ("input1_mm" and "input2_mm") located in the "Mol_MM" sub-directory (all these files are automatically generated by the R.E.D. III program) have to be uploaded (see Pictures 49 to 51).
 |
 |
 |
CONCLUSION
This tutorial example demonstrated the project submission to R.E.DD.B. of ten whole molecules. The "Summary of information" corresponding to this project can be obtained from "here". The data corresponding to this project can also be downloaded using the "Download projects" R.E.DD.B. tool available from the R.E.DD.B. main page.
-I.3- Update of projects in R.E.DD.B.
A project available in R.E.DD.B. can be updated by its author any time after it has been recorded in R.E.DD.B. (see FAQ-10). When updating a R.E.DD.B. project, two different types of information can be added to a R.E.DD.B. project:
- The publication reference corresponding to a R.E.DD.B. project can be provided once it is known. Indeed, it might be more suitable to submit a project to R.E.DD.B. before publication (see the FAQ-3 for more information). This means that the publication reference is not provided during the project submission to R.E.DD.B.
- The five force field related files, that the author of a R.E.DD.B. project has the freedom to upload (or not upload) during the project submission to R.E.DD.B., can be added to this project any time she/he wishes.
The "Update project" R.E.DD.B. tool is available from the R.E.DD.B. main page (see Picture 52). Using this tool and to update one of her/his R.E.DD.B. project, an author has to provide her/his R.E.DD.B. login and password received by email after registration, and click on the "OK" button (see Picture 4).
 |
After providing a login and password, a new page entitled "UPDATE A PROJECT 1/4" is automatically opened (see Picture 53).
 |
The tasks performed by the "Update projects" R.E.DD.B. tool are reminded (see Picture 54).
 |
An Author has to select among all the projects she/he has already submitted, the project she/he wishes to update, and then clicks on the "OK" button (see Picture 55). An author has obviously the rights to update only the R.E.DD.B. project(s) she/he has submitted to R.E.DD.B. Let us select an hypothetical "W-1" R.E.DD.B. project, as an example.
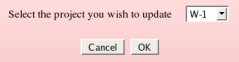 |
A new page entitled "UPDATE A PROJECT 2/4" is then automatically opened. The R.E.DD.B. project being updated is reminded: its R.E.DD.B. code is displayed (see Picture 56).
 |
The publication reference was not provided during the project submission to R.E.DD.B. since unknown at the time of the submission. One has the possibility to provide it if one wishes (see Picture 57a). Let us decide to provide the publication reference by toggling on the "YES" button (see Picture 57b).
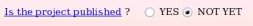 | => | 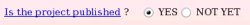 |
None of the force field related files compatible with the "W-1" R.E.DD.B. project were provided during the project submission to R.E.DD.B. Indeed, the primary goal of R.E.DD.B. is to provide RESP or ESP charge values for new molecules or molecule fragments, and not to give force field related information (see Picture 58a). However, it could be convenient to provide force field information (see FAQ-7; § -7.3-) compatible with a R.E.DD.B. project when needed. For instance, the corresponding files could be required by a user after discussion in a mailing list, and provided to the community by updating the corresponding R.E.DD.B. project. Let us decide to only provide the force field related file number 1, i. e. the file allowing the convertion of a Tripos mol2 file into an AMBER OFF force field library, by toggling on the "YES" button corresponding to the "File 1" (see Picture 58b).
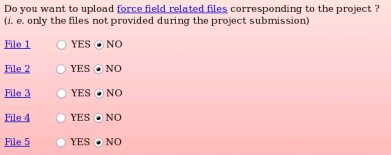 | => | 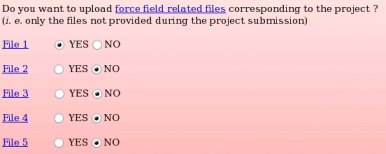 |
When ready one clicks on the "OK" button (see Picture 59).
 |
A new page entitled "UPDATE A PROJECT 3/4" is then automatically opened. The R.E.DD.B. project being updated is reminded: its R.E.DD.B. code is displayed (see Picture 60).
 |
The publication reference is provided (see Picture 61).
 |
The LEaP script allowing the conversion of the Tripos mol2 file format into an AMBER OFF force field library is provided. The upload of this file is performed by browsing the file system tree and selecting the corresponding file (see Picture 62).
 |
When ready one clicks on the "OK" button to record the information provided [or on the "Previous" button to reload the previous update page and perform some corrections of the choices made (see Picture 63)].
 |
A new page entitled "UPDATE A PROJECT 4/4" is then automatically opened. The R.E.DD.B. project being updated is reminded: its R.E.DD.B. code is displayed (see Picture 64).
 |
The publication reference has been recorded in R.E.DD.B. (see Picture 65).
 |
 |
The force field related file number 1 has been downloaded (see Picture 66).
 |
The update project procedure for the current project is now done. One can close the window, or update a new project.
REMARKS
- It is important to underline that, when one updates a project, the date of update is automatically recorded in R.E.DD.B., and a new "Summary of information" for this project is generated with the date of update clearly displayed (see Picture 67). An example of updated "Summary of information" is available for the "W-46" R.E.DD.B. project. It can be obtained by clicking from "here".
 |
 |
- Finally, a project that has been updated a first time can be re-updated. Thus, for the project update example just described, the publication reference has been provided, and the force field related file number 1 has been uploaded. Consequently only the upload of the force field related file numbers 2 to 5 are going to be allowed if one decides to update this project a second time. Thus, if the publication reference is already known and the five force field related files have been already uploaded during the project submission procedure or during a first update, the corresponding project can not be updated anymore, and specific messages are printed in this case (see Picture 68).
 |
-I.4- List and download of R.E.DD.B. projects
One does not need to be registered as a R.E.DD.B. user to search and download projects in R.E.DD.B. (see FAQ-10 for more information). The projects available in R.E.DD.B. are free for download for the academics and non-academics users .
-I.4.1- List of R.E.DD.B. projects
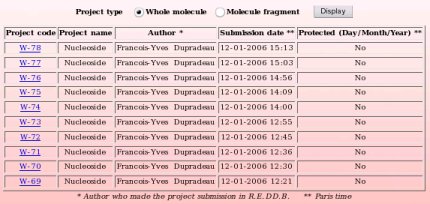
The R.E.DD.B. browser or "List projects" R.E.DD.B. tool available from the R.E.DD.B. main page provides a quick and convenient way for displaying R.E.DD.B. projects, the latters being classified into two categories: "whole molecule" and "molecule fragment" projects. The projects diplayed fulfill the following rule: The last projects submitted to R.E.DD.B. are first displayed (see Picture 69).
 |
 |
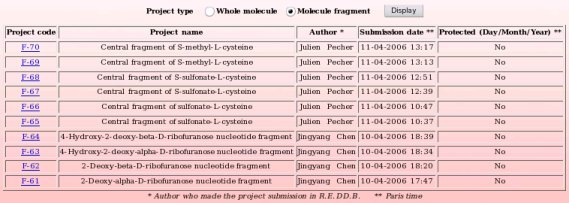
By toggling on the "Whole molecule" or "Molecule fragment" button, and clicking on the "Display" button, one displays either the whole molecule or molecule fragment projects recorded in R.E.DD.B. (see Pictures 70a and 70b, respectively). R.E.DD.B. projects are displayed by blocks of ten projects.
 |  | |
By clicking on the "left red arrow" one has access to R.E.DD.B. projects anterior to the block of ten projects displayed. By clicking on the "right red arrow" one has access to R.E.DD.B. projects posterior to the block of ten projects displayed (see Picture 71). By clicking on the "Download projects" link, one opens the "Download projects" R.E.DD.B. tool also available from the R.E.DD.B. main page.
 |
By clicking on a R.E.DD.B. code, a popup window is opened. Let us click on the "F-70" R.E.DD.B. code, for instance. The new window represents an "Extract of information" for the current project (see Picture 72).
 |
The complete "Summary of information" of the "F-70" project is available by clicking on the "click here" link (see Picture 73).
 |
The "Extract of information" contains the project keywords and the whole molecule and molecule fragment names available in the project (see Picture 74). By giving information about projects, the "Extract of information" provides a starting point for the donwload of the R.E.DD.B. project(s) if one is interested in this project.
 |
 |
-I.4.2- Download of R.E.DD.B. projects
To search and then download a R.E.DD.B., one has to use the "Download projects" R.E.DD.B. tool available from the R.E.DD.B. main page. After clicking on the corresponding link, a new window is opened (see Picture 75).
 |
By clicking on the "Corrections applied to R.E.DD.B. after its release" link, one has access to a page which summarizes the corrections applied to the R.E.DD.B. projects after their official submission date (see Picture 76). Thus, before to download a project from R.E.DD.B. it is a good idea to check for the errors found in R.E.DD.B projects.
 |
The "Download projects" R.E.DD.B. tool offers five different ways to search in R.E.DD.B. (see FAQ-10 for more information). One can search by 'Project code', 'Molecule keyword', 'Molecule name', by 'Author lastname' and by 'Theory level/Basis set' [the searches being independent of the character case (upper or lower), the default search method being by 'Molecule name']. The query is entered in the Text box, and the search is automatically performed after clicking on the "OK" button (see Picture 77). The search results are then displayed in the same window, below the search box.
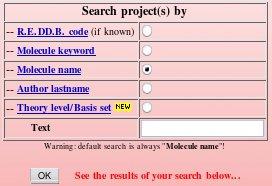 |
Let us perform an example of search using the "W-46" project code (see Picture 78). The exact R.E.DD.B. code has to be provided to be found.
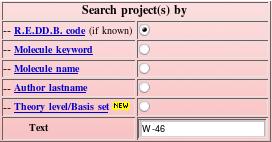 |
To display the results of the search one has to use the scroll bar to move down the window. The search has been performed, and is successfull (the word "[Done]" is printed). The name of the R.E.DD.B. project is then diplayed, and the "Summary of information" of the project is available by clicking on the "Click Here" link (see Picture 79).
 |
Each Tripos mol2, PDB and fitting program input file belonging to the "W-46" R.E.DD.B. project can be individually downloaded by clicking on the corresponding link (see Picture 80).
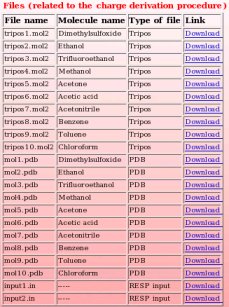 |
Each force field related file belonging to the the "W-46" R.E.DD.B. project (that the author has the freedom to provide during the project submission procedure) can also be individually downloaded (see Picture 81).
 |
A single ".tar.bz2" file containing all the files of the R.E.DD.B. project can also be downloaded (see Picture 82). The project files are archived with the tar program, and compressed with the bzip2 program.
 |
REMARKS
- When performing a search in R.E.DD.B. using a 'Project code' or the 'Author lastname', the exact code and name have to be provided to lead to a positive patch. On the contrary, searching in R.E.DD.B. using a shorter 'Molecule keyword' or 'Molecule name' is authorized (see FAQ-10 for more information). Such general search can be usefull when one is interested in searching a familly of projects. Thus for instance, performing a search using the "Acet" fragment of 'Molecule name' leads to many positive matches (Project names = "N-Methylacetamide", "Acetonitrile", "Organic solvent", "N-Acetyl-L-alanine-N'-methylamide", "Central fragment of alpha-aminoisobutyric acid", "N-terminal charged fragment of alpha-aminoisobutyric acid", "C-terminal charged fragment of alpha-aminoisobutyric acid", "N-terminal neutral fragment of alpha-aminoisobutyric acid", "C-terminal neutral fragment of alpha-aminoisobutyric acid", "Central fragment of O-methyl-L-tyrosine", "N-terminal charged fragment of O-methyl-L-tyrosine", "C-terminal charged fragment of O-methyl-L-tyrosine", "N-terminal neutral fragment of O-methyl-L-tyrosine", "C-terminal neutral fragment of O-methyl-L-tyrosine").
- When performing a search, protected R.E.DD.B. projects (see FAQ-4 for more information) are found, but their corresponding data can not be downloaded during the period of protection (see Picture 83).
 |
Should you find any mistake in this tutorial, please, send me an e-mail:
If you have questions about this tutorial, please, send your emails to the q4md-forcefieldtools mailing list. We will answer to the queries about the q4md-forcefield tools in the Amber or CCL mailing lists as well.
Last update of this tutorial page: December 10th, 2015.
Charge derivation data free for download.
Université de Picardie Jules Verne. Sanford Burnham Prebys Medical Discovery Institute.
© 2009-2024. All rights reserved.
