Ante_R.E.D.-1.x & R.E.D. III.x programs
F.-Y. Dupradeau *
Université de Picardie - Jules Verne, Amiens
P. Cieplak
Sanford Burnham Prebys Medical Discovery Institute, La Jolla, CA
Information about this tutorial can be found in the following reference:
P. Cieplak, W.D. Cornell, C. Bayly & P.A. Kollman, Application of the multimolecule and multiconformational RESP methodology to biopolymers: Charge derivation for DNA, RNA, and proteins. J. Comput. Chem. 1995, 16, 1357-1377, Abstract, [Local PDF file].
This tutorial presents the execution of the Ante_R.E.D.-1.x and R.E.D. III.x programs to derive RESP charges for (i) whole molecules and (ii) molecule fragments using the dimethylalanine (AIB, 1-aminoisobutyric acid) dipeptide as example. RESP charge derivation involving $n molecules, $i conformations and $j orientations are described in a sequential approach.
Although eight different charge derivation procedures have been developed in the R.E.D. III.x program, it is important to underline that only RESP charge derivations useful in simulations based on the Cornell et al. force field (and its different adaptations) are demonstrated in this tutorial. Many examples and choices made are described, and whole data generated from R.E.D. III.x runs are available for download. However, RESP charges required for force field simulations using the GLYCAM force fields, and ESP charges used in some CHARMM, OPLS and even AMBER force field simulations can also be generated following similar strategies to those presented. Moreover, by changing few parameters in the R.E.D. source code, an infinite number of different procedures can be developed. Thus for instance, by modifying a single line in the R.E.D. III.x source code, charge values compatible with the Duan et al. force field can also be derived.
This tutorial has been tested on a laptop with a single PIV 2.4 GHz cpu and with 1 Gb RAM under Linux Fedora Core 4.0 (kernel 2.6.12.3) using the 22 NOV 2004 (R1) GAMESS version, the Gaussian 1998 RevA.11.4 version and the RESP version from AMBER8 (RESP was recompiled using qtol = 0.1d-5, maxq = 5000, maxlgr = 500 and maxmol = 200).
This tutorial has been updated in agreement with the new features incorporated in R.E.D. III.4 and R.E.D. IV version June 2010.
RESP charge derivation using the Ante_R.E.D.-1.x & R.E.D. III.x programs
-I.1- A "Mini HowTo"
-I.2- General information about input files required by R.E.D. III.x
-I.2.1- The initial P2N file
-I.2.2- The Ante_R.E.D. program
-I.3- The tutorial itself
-I.3.1- Preparation of the R.E.D. III.x inputs
-I.3.2- Execution of R.E.D. III.x
-I.3.2.1- Single-conformation single-orientation RESP fits for new dipeptide molecules
-I.3.2.2- Single- or multi-conformation multi-orientation RESP fits for new dipeptide molecules
-I.3.2.3- Single- or multi-conformation multi-orientation RESP fits for the central fragment of new amino-acids
-I.3.2.4- Multi-molecule multi-conformation multi-orientation RESP fits for terminal fragments of new amino-acids
-I.3.2.4.1- Implementation of inter-molecular charge constraints & inter-molecular charge equivalencing in R.E.D. III.x
-I.3.2.4.2- Back to the tutorial
-I.3.2.5- Multi-molecule multi-conformation multi-orientation RESP fits for other fragments
-I.3.3- The Tripos mol2 file format
-I.3.3.1- Advantages of the Tripos mol2 file format
-I.3.3.2- Why do we use the Tripos mol2 file format in R.E.D.
-I.3.3.3- Using the Tripos mol2 file format to visualize structures and charge values within an Internet Browser
-I.3.3.4- Using the Tripos mol2 file format as library precursor for AMBER or CHARMM force field library
-I.3.2.4.1- Whole molecule case
-I.3.2.4.2- Molecule fragment case
-I.3.3.5- Using Tripos mol2 files in LEaP: Implementation of new features in LEaP
-I.3.4.- The mol3 file format
RESP charge derivation using Ante_R.E.D.-1.x & R.E.D. III.x
For a new molecule, RESP and ESP charge derivation is performed in three steps (i) Geometry optimization, (ii) Molecular Electrostatic Potential (MEP) computation using the optimized geometry obtained in the first step and (iii) fitting the charges centered on the atoms to the MEP calculated in the second step. The RESP ESP charge Derive program (R.E.D.) sequentially executes these three steps by interfacing the GAMESS (GAMESS-US or Firefly) or Gaussian (Gaussian 94, 98 or 03 version) quantum mechanics (QM) program and the RESP program, and allows the automatic derivation of RESP and ESP charges for the target molecule.
With the R.E.D. version I, highly reproducible RESP and ESP charge values (+/- 0.0001 e) can be derived by controlling the molecular orientation of the optimized geometry of a single conformation of a single molecule whichever the QM program or initial structure choice is. A new charge derivation procedure has been developed based on multi-orientation RESP or ESP fit. The use of multiple molecular orientations in the fitting process is supposed to limit the charge uncertainty induced when using a single orientation. With the R.E.D. version II, the use of multiple conformations of a single molecule in the fitting procedure has been implemented allowing for the derivation of charge values of high quality (i. e. more general; needed in molecular dynamics simulation). Multi-conformation and multi-orientation RESP or ESP fit can be performed together or independently for structures containing chemical elements of the periodic table up to Z = 35 (Bromine) according to the user's choice. With the R.E.D. version III, the control of charge constraints for atoms and groups of atoms in a molecule or a set of molecules (intra-molecule charge constraint) or between two molecules (inter-molecule charge constraint and inter-molecular charge equivalencing) has been developed allowing for the derivation of the RESP and ESP atom charge values for molecule fragments and sets of molecules. Thus, fitting procedures involving multiple molecules, and for each molecule, multiple conformations, and for each conformation, multiple orientations, can now be automatically carried out. Finally, eight different charge derivation procedures using different MEP computation algorithms (Connolly surface and CHELPG algorithms) and different fitting procedures (with or without hyperbolic restraints) are now available. Potentially, an infinite number of approaches can be developed by simply changing a few words in the R.E.D. III.x source code. Such procedures can be used in simulations based on AMBER, CHARMM, OPLS and GLYCAM force fields. Once the R.E.D. execution has correctly completed, the charge values are available in Tripos mol2 file(s) which can be considered as precursors of AMBER OFF and CHARMM RFT or PSF force field libraries.
-I.1- A "Mini HowTo"
The global procedure for deriving RESP or ESP charge values and building force field library(ies) for new molecules and/or new molecular fragments is summarized in Figure 1:
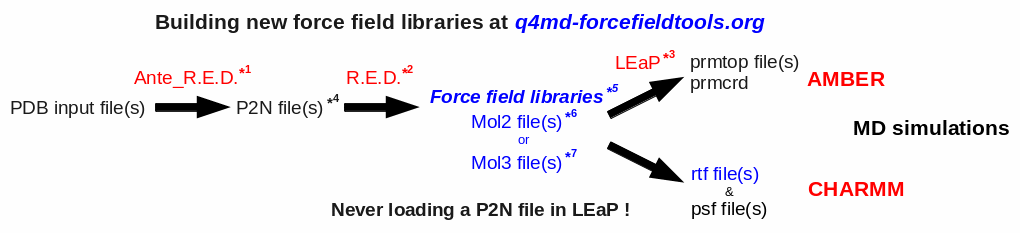
| *1 Ante_R.E.D. 1.x from the R.E.D. III.x tools or Ante_R.E.D. 2.0 at R.E.D. Server | *5 Important features of a force field library: | |
| *2 R.E.D. III.x from the R.E.D. III.x tools or R.E.D. IV at R.E.D. Server | - Chemically equivalent atoms bear the same charge value | |
| *3 tLEaP or xLEaP (handle force fields & force field libraries): | - Two atoms belonging to a residue cannot share the same name | |
| - Two atoms sharing the same name in a given residue are not recognized by LEaP ! | *6 Description of the Tripos mol2 file format here and here | |
| - Force field atom types are added by using a LEaP script (see an example here) | *7 Description of the mol3 file format here | |
| *4 A P2N file is a PDB file with two columns of atom names: | ||
| Main characteristics (more information here, and an example here): | ||
| - 1st column of atom names reflecting chemical equivalencing (chemically equivalent atoms bear the same atom name; yellow column here) | ||
| - 2nd column of atom names reflecting IUPAC naming conventions (two atoms belonging to a residue cannot share the same name; red column here) |
Below is a "Mini HowTo" that summarizes the important steps one could follow in order to derive RESP or ESP atom charge values for whole molecule(s) and/or molecule fragment(s) starting from $n molecules, $i conformations and $j orientations using the Ante_R.E.D. and R.E.D. III.x programs.
-1- Build the $n initial whole molecules in a graphical interface or some other tools, and save them in the Protein Data Bank (PDB) format. When different conformations of a molecule are used in charge fitting, generate the conformers separately, and save them in the PDB format.
-2- Carefully check the atom names, residue names and residue numbers in each of these PDB files since they will be used in the Tripos mol2 files (or, in other words, the force field library precursors) generated by R.E.D. III.x, and in AMBER OFF and CHARMM RTF/PSF force field libraries. Indeed, when one has the goal of developing a new force field library two different atoms in a PDB file cannot have the same atom name within a single residue, and a residue is characterized by a single residue name and a single residue number. Moreover, two residues differ at least by their residue number. Finally, at this stage, the presence of the atom connectivities in the PDB files and the atom order of a molecule to the PDB format do not really matter (although the atom order of two conformations of a molecule must be the same in each PDB file, when multi-conformational calculations are performed).
-3- Execute the Ante_R.E.D. program to convert the different PDB files into the corresponding P2N files by repeating the following command for each PDB file:
perl Ante_RED.pl File.pdb
or
perl Ante_RED.pl File.pdb > Ante_RED.log
-4- Check and modify the P2N files generated by Ante_R.E.D. (an example of P2N file is available by clicking here).
- For each molecule to the P2N format, determine its name (using the international nomenclature rules for instance), and add it to each P2N file as the molecule title (Example: "REMARK TITLE Dimethylalanine-dipeptide"). This molecule name is used in the GAMESS, Gaussian, and RESP input files during the charge derivation procedure, and is required in the corresponding project submission in the RESP ESP charge DDataBase (if one wishes to use this option).
- Check whether the molecule total charge value (the default value is 0.0; Example: "REMARK CHARGE-VALUE 0") and the electronic spin multiplicity (the default value is 1.0; Example: "REMARK MULTIPLICITY-VALUE 1") generated by Ante_R.E.D. are correct. If not, provide the correct values.
- Check the atom connectivities whether they are all listed and correct. Those connectivities are crucial because they are used to create molecular topology information in the Tripos mol2 files generated by R.E.D. III.x, but also in the AMBER and CHARMM force field libraries.
- Select the re-orientation procedure (i. e. QMRA or RBRA) which will be applied to the optimized Cartesian coordinates for each of the molecules used in the charge derivation procedure. This involves the use of a single molecular orientation or multiple orientations in the RESP or ESP fit, respectively. In the case of selecting the RBRA approach, add the corresponding keyword (Example: "REMARK REORIENT 5 18 19" or "REMARK REORIENT 5 18 19 | 19 18 5"). With R.E.D. III.4 and R.E.D. IV version June 2010, two new transformation procedures have been developed: they differentiate translation from rotation (see below).
- Check the atom names found in each P2N file. PDB and P2N files only differ by the presence of a single and two columns of atom names, respectively. In a P2N file, the first column of atom names (or "yellow column" examplified in the P2N file below) is used in the automatic RESP input generation, which was originally implemented in the R.E.D. program version I. The rules followed by R.E.D. to generate these RESP inputs are defined below. The second column (or "red column" also examplified in the P2N file below) contains the original molecule atom names (defined in the PDB file before Ante_R.E.D. execution), and are those used in the Tripos mol2 files generated by R.E.D. III.x (and consequently in the AMBER and CHARMM force field libraries). In the absence of such a second column, the atom names found in the Tripos mol2 files are automatically generated by R.E.D., and are constituted by the chemical symbol of the chemical element and a number which is incremented.
- Add the keywords corresponding to the use of intra-molecular charge constraint(s) (Example: "REMARK INTRA-MCC 0.0 | 1 2 3 4 5 6 | R" or "REMARK INTRA-MCC 0.2719 | 8 | K"), inter-molecular charge constraint(s) (Example: "REMARK INTER-MCC 0.0 | 1 2 | 1 2 3 4 | 1 2 3 4 5 6 7 8"), and/or inter-molecular charge equivalencing (Example: "REMARK INTER-MEQA 1 2 3 4 | 1 2 3 4 5 6 7 8") if one wishes to use such constraints during a RESP or ESP fit. Such constraints are required in charge derivation of molecule fragments and force field topology databases.
- Check the atom order, especially, whether the hydrogen atoms are located after their heavy atom counterparts (quickly locating methylene and methyl groups in a molecule is convenient in RESP charge derivation, in particular when one wishes to study the corresponding RESP inputs).
- Finally, in the case of multi-conformation charge fitting, merge the P2N files of the different conformations (each single P2N file should have the same atom order) of a molecule into a single P2N file. In this file, each set of cartesian cordinates representing a conformation are separated by the 'TER' keyword, and a single set of atom connectivities is conserved and located after the first set of Cartesian coordinates.
-5- Rename (or create a symbolic link for) each of the $n P2N files generated by Ante_R.E.D. for the $n molecules into $n Mol_red$n.p2n R.E.D. III.x input files.
-6- Decide whether one wishes to run R.E.D. III.x using the $OPT_Calc = "On" variable, or using the $OPT_Calc = "Off" one (this parameter can be changed in the main program of the R.E.D. III.x perl script).
- If $OPT_Calc = "Off" is setup in R.E.D. III.x, then the geometry optimization for each of the $n molecules (and for the different conformations of each molecule, if required) should be run separately in a standalone mode i. e. using the Gaussian or GAMESS input(s) that are automatically generated by Ante_R.E.D. If different conformations are used for a molecule in a multi-conformational charge fit, concatenate the geometry optimization output files of these conformations into a single file. Finally, rename (or create a symbolic link for) the Gaussian or GAMESS outputs of the $n molecules into $n Mol_red$n.log files, keeping the order of the $n Mol_red$n.log files identical to that of the $n Mol_red$n.p2n P2N files. Run R.E.D. III.x by using the following command:
perl RED-vIII.4.pl
or
perl RED-vIII.4.pl > RED-vIII.log
- If $OPT_Calc = "On" is setup in R.E.D. III.x, then simply run R.E.D. III.x using the command as just above ("perl RED-vIII.4.pl > RED-vIII.log"). Althought it seems simpler, such approach is not recommended when deriving RESP or ESP charge values using a multiple-molecule multiple-conformation and/or multiple-orientation RESP fit. This is because it is unlikely that all the geometry optimization jobs for the different molecules and conformations converge. In the latter case, this leads to a "FAILED" R.E.D. III.x execution.
-7- Convert the Tripos mol2 files generated by R.E.D. III.x into force field libraries specific of AMBER or CHARMM using adequate scripts or programs. For AMBER, LEaP scripts can be written to convert Tripos mol2 files into OFF libraries (examples of such LEaP scripts are already available in the RESP ESP charge DDataBase; click here and here for examples). The convertion of Tripos mol2 files into CHARMM force field libraries will be handled in the future version of R.E.D. However, a project available in the RESP ESP charge DDataBase can be updated and a script dedicated to this task can be added to a project by its author any time she/he wishes (click here and here for examples).
-8- Submit the data generated by R.E.D. III.x into the RESP ESP charge DDataBase or R.E.DD.B. (if one wishes to use this option).
-I.2- General information about input files required by R.E.D. III.x
-I.2.1- The initial P2N file
To execute the R.E.D. program version III an initial P2N file (PDB file format with two columns of atom names and the P2N extension) for _each molecule_ used in the charge derivation procedure is required (previously a single initial PDB file was used by R.E.D. II). R.E.D. III.x automatically recognizes P2N files having the 'Mol_red$n.p2n' file name(s) (where $n is the number of molecule(s) used in the fitting procedure; the "$MOL_START" variable available in the R.E.D. II source code is obsolete in R.E.D. III.x). In this file the following information has to be provided: the molecule title, the molecule total charge, the spin multiplicity of the molecule, the Cartesian coodinates of each conformation describing the molecule, the information about the molecule orientation(s) used in the MEP computation, the atom connectivities, the residue name(s) and the atom names (1 or 2 columns of atom names). Information about intra-molecular charge constraint(s) within a molecule, inter-molecular charge constraint(s), and/or the inter-molecular charge equivalencing between different molecules are also required in charge derivation of molecule fragments and force field topology databases.
-1- The molecule title is defined using the "REMARK TITLE" keywords (Example: REMARK TITLE Dimethylalanine-dipeptide; do not use any space character with the title itself; keywords present at the beginning of a line). If the title is not provided the default title 'Molecule_$n' is used ($n is the number of molecule(s) used in the fitting procedure, the "$TITLE" variable available in the R.E.D. II source code is obsolete). The molecule title is used in the three steps that R.E.D. sequentially executes (geometry optimization, MEP computation and fitting inputs).
-2- The total charge of the molecule is defined using the "REMARK CHARGE-VALUE" keywords (Example: REMARK CHARGE-VALUE 0; keywords present at the beginning of a line). If the total charge value is not found, the default value of 0.0 is assigned (the "$CHR_VAL" variable available in the R.E.D. II source code is obsolete). The total charge of the molecule is required in the geometry optimization and MEP computation steps.
-3- The spin multiplicity of the molecule is defined using the "REMARK MULTIPLICITY-VALUE" keywords (Example: REMARK MULTIPLICITY-VALUE 1; keywords present at the beginning of a line). If the spin multiplicity value is not provided, the default value of '1' is assigned (the "$MLT_VAL" variable available in the R.E.D. II source code is obsolete). The spin multiplicity of the molecule is required in the geometry optimization and MEP computation steps.
-4- The Cartesian coordinates of each conformation describing the molecule have to be provided and have to be separated with the "TER" keyword. The atom order in each conformation has to be the same (this was implemented in R.E.D. II and was not modified in R.E.D. III.x).
-5- The residue name(s) and residue number(s) have to follow rules 'that make sense'. In particular, a single residue name and residue number must characterize a residue: The residue name(s) available in an initial P2N file is(are) used by R.E.D. for the definition of the residue name(s) in a Tripos mol2 file (this was implemented in R.E.D. II and was not modified in R.E.D. III.x). This residue name also plays an important role in the AMBER OFF and CHARMM RFT/PSF force field library definition. The "UNK" residue name used by many graphic interfaces is a generic name which is not representative of the studied molecule. Consequently, "UNK" should be replaced by a more specific residue name.
-6- The atom connectivities have to be provided (only for the first conformation of a molecule since the atom order is identical in different conformations of a molecule) if the user wants the R.E.D. program to generate corresponding Tripos mol2 file(s) at the end of the R.E.D. execution (this was implemented in R.E.D. II and was not modified in R.E.D. III.x).
-7- The information about the molecular orientation of each optimized geometry used in the MEP computation (i. e. the choice of the re-orientation procedure applied before the MEP computation) has also to be provided.
General information about molecular orientation and charge values
It is known that the number of MEP points depends on the molecular orientation in space. Consequently, the values of derived RESP and ESP atom charges also depend on the molecular orientation of the optimized geometry. The molecular orientation can be partially controlled in the GAMESS program using the molecular principal axes ("COORD = CART" keyword) and in the Gaussian program by placing the center of nuclear charge at the origin ("Symmetry" keyword). Thus, since GAMESS and Gaussian do not apply the same internal algorithm, the molecular orientation of each optimized structure generated by both programs will be different. Moreover, each QM program can generate different molecular orientations for a target minimum using its internal re-orientation algorithm. To address these problems, a new reorientation algorithm has been introduced in R.E.D. version I which can be applied to the optimized molecular system before calculating the MEP. It allows a full control of the molecular orientation of the optimized geometry independently of the QM program or initial structure selected. Consequently, highly reproducible RESP or ESP charges can be derived. This procedure applied to the optimized geometry uses a Rigid-Body Re-orientation Algorithm based on the choice of three atoms. The first two atoms define the (O, X) axis, the third one defines the (O, X, Y) plane; the Z-axis being defined as the X x Y cross-product.
Different procedures for controlling the molecular orientation of the optimized geometry with R.E.D.
Three different procedures are available in R.E.D. to control the molecular orientation of the optimized geometry before computing the MEP. We strongly recommend to use the third one in RESP or ESP charge derivation. With R.E.D. III.4 and R.E.D. IV version June 2010, two additional transformation procedures have been developed. They differentiate translation from rotation (see below):
-1- If re-orientation information based on three atoms (see case -2-, below) is not provided in the initial P2N file, then the internal Re-orientation Algorithm available in GAMESS or Gaussian is used to reorient the optimized Cartesian coordinates (we called this the QMRA procedure). In this case, the molecular orientation of the optimized geometry is partially controlled, the charges derived using the two QM programs differ from each other, and are not reproducible. Indeed, it is important to underline that the Gaussian program reorients by default (using its own internal algorithm by placing the center of nuclear charge at the origin) a Cartesian coordinate set each time a molecular energy calculation is performed. This leads to the well known set of coordinates called 'Standard orientation'. Thus, at the end of a geometry optimization the Cartesian coordinates generated by Gaussian are always re-oriented with respect to the initial geometry (if the "Symmetry" keyword is used in the input). On the contrary, the GAMESS program only reorients the molecular Cartesian coordinates once at the beginning of a job (if the "COORD = CART" keyword is used). Thus, at the end of a geometry optimization the Cartesian coordinates generated by GAMESS are not perfectly re-oriented (according to the GAMESS internal algorithm). In order to get a well re-oriented optimized geometry using GAMESS, one has to use the optimized Cartesian coordinates in a new single point energy run. In the R.E.D. I version, this was performed during the MEP computation (if the Rigid-Body Re-orientation Algorithm was not used). This has been modified in the R.E.D. II version, where a new routine has been incorporated (which uses the Jacobi matrix diagonalization method) to re-orient the GAMESS optimized geometry. This is a similar algorithm to the one used internally by GAMESS, and presents the advantage that the GAMESS type molecular orientation can be generated without the need of executing the GAMESS program itself. Finally, such a 'standalone' re-orientation algorithm is not needed if the Gaussian program is used since the molecular orientation available at the end of the geometry optimization output is well re-oriented (according to the Gaussian internal algorithm).
-2- If re-orientation information based on three atom numbers (not atom names!) is provided in the initial P2N file, the optimized geometry is automatically re-oriented using the Rigid-Body Re-orientation Algorithm implemented in R.E.D. (we called this the RBRA procedure). Thus, in this case the molecular orientation of the optimized geometry is fully controlled and highly reproducible RESP and ESP charges can be derived independently of the QM software or initial structure choice. The charges are reproducible since the three atoms used in the RBRA procedure are known. In order to apply this approach, one has to provide the following keywords in the P2N file format before the set of Cartesian coordinates of the first conformation:
After the "REMARK REORIENT" keywords (which has to be present at the beginning of a line), one needs to provide the atom numbers involved in the Rigid-Body Re-orientation Algorithm i. e.
REMARK REORIENT atm_nb$A atm_nb$B atm_nb$C where "$A", "$B" and "$C" stand for the atom numbers.
-3- If $j sets of three atom numbers [$j is a positive integer representing the number of molecular orientation(s); an infinite number of orientations can be in principle used] are given, the optimized Cartesian coordinates obtained from the QM program output are reoriented $j times using the Rigid-Body Re-orientation Algorithm (we called this the multi-RBRA procedure). The reoriented sets of Cartesian coordinates are then used to compute $j MEPs. A $j orientation fit (or multi-orientation RESP fit) is then applied using the RESP program. This allows averaging out the charge value differences observed for a particular orientation over several different orientations. The derived atom charges are also highly reproducible in this case. Here is an example in which $j = 4:
REMARK REORIENT atm_nb$A atm_nb$B atm_nb$C | atm_nb$C atm_nb$B atm_nb$A | atm_nb$D atm_nb$C atm_nb$A | atm_nb$A atm_nb$C atm_nb$D
- The keywords and the atom numbers must be written in the same line,
- The pipe character "|" is used as separator between two different orientations,
- "$A", "$B", "$C", and "$D" are the atom numbers.
For larger numbers of molecular reorientations, the corresponding format can be used, each line starting by the "REMARK REORIENT" keywords i. e. using several lines:
REMARK REORIENT atm_nb$A atm_nb$B atm_nb$C | atm_nb$C atm_nb$B atm_nb$A
REMARK REORIENT atm_nb$D atm_nb$B atm_nb$A | atm_nb$A atm_nb$B atm_nb$D
REMARK REORIENT atm_nb$E atm_nb$D atm_nb$A | atm_nb$A atm_nb$D atm_nb$E
General remarks
As mentioned above, the R.E.D. III.x program can be used to derive RESP or ESP charges for $n molecules, and for each molecule $i conformations, and for each conformation $j molecular orientations. For each molecule represented by a P2N file and used in the charge derivation procedure, the three molecular re-rorientation procedures previously described to control the molecular orientation of an optimized geometry can be independentyl used to perform charge fitting:
- For a molecule, if the information about the Rigid Body Re-orientation Algorithm information is not provided in its initial P2N file, a $i conformation RESP fit is performed using the molecular orientation calculated by the internal Re-orientation Algorithm of the QM program (QMRA procedure).
- For a molecule, if $j orientations ($j = 1 or a higher positive integer, denoting the molecular orientation based on the Rigid-Body Re-orientation Algorithm) are requested, a $i conformation * $j orientation RESP fit is performed (RBRA or multi-RBRA procedure). In this case, the re-orientation information based on the three atom names is only provided for the first conformation (before the first set of Cartesian coordinates) and is used for the $i conformations.
- R.E.D. III.x allows using the geometry optimization output (single conformation optimization output or concatenated file created after $i conformation optimizations) generated by the GAMESS program as input for MEP computation using the Gaussian program (and vice-versa). If the Rigid-Body Re-orientation Algorithm is not selected, the molecular orientation used to compute the MEP is based on the QM output and is not based on the QM software. This means that if a Gaussian geometry optimization output is used to compute MEP using the GAMESS program (for instance), then the molecular orientation selected is based on the Gaussian internal re-orientation algorithm. However, this particularity is obviously avoided if the Rigid-Body Re-orientation Algorithm is used.
Two additional transformation procedures have been implemented in R.E.D. III.4 and R.E.D. IV version June 2010
These new procedures allow differentiating translation from rotation. In the RBRA procedure presented in the case -2- and case -3- above, the first selected atom is translated to the origin of axes, the first two atoms define the (O, X) axis while the third one is used to define the (O, X, Y) plane. The (O, Z) axis is automatically set as the cross-product between the (O, X) and (O, Y) axes. Thus, the RBRA algorithm uses translations and rotations to reorient the considered geometry. As charge values are affected by the RBRA procedure, this raises a new question: are translation and/or rotation responsible of these charge discrepancies? To answer to this question two new transformation procedures have been developed: the first one uses only translation(s), while the second one corresponds to rotations.
-4- Rotation
Rotation and multiple-rotations can be carried out using the "REMARK ROTATE" keywords. After the "REMARK ROTATE" keywords (which has to be present at the beginning of a line), one needs to provide the atom numbers involved in the rigid-body rotation algorithm. The following format has to be followed:
REMARK ROTATE atm_nb$A atm_nb$B atm_nb$C, where "$A", "$B" and "$C" stand for the atom numbers.
By analogy to the multiple re-orientation procedure described above (case -3-), multiple-rotations can be carried out (the pipe character "|" can be used as separator between two different rotations):
Examples of multiple-rotations:
REMARK ROTATE atm_nb$A atm_nb$B atm_nb$C | atm_nb$C atm_nb$B atm_nb$A, or
REMARK ROTATE atm_nb$A atm_nb$B atm_nb$C
REMARK ROTATE atm_nb$C atm_nb$B atm_nb$A
-5- Translation
Translations and multiple-translations can be carried out using the "REMARK TRANSLATE" keywords. After the "REMARK TRANSLATE" keywords (which has to be present at the beginning of a line), one needs to provide three values for the rigid-body translations one wants to perform on the X, Y and Z axes. The following format has to be followed:
REMARK TRANSLATE $dX $dY $dZ where "$dX", "$dY" and "$dZ" stand for the translations done on the X, Y and Z axes.
Example:
REMARK TRANSLATE 1 0 -1.5 means that +1 is added to the X Cartesian coordinates, and -1.5 is substracted to the Z axis for the selected orientation (while Y Cartesian coordinates remain unaffected).
By analogy to the multiple re-orientation procedure described above (case -3-), multiple-translations can be carried out (the pipe character "|" can be used as separator between two different translations):
Examples of multiple-translations:
REMARK TRANSLATE $dX1 $dY1 $dZ1 | $dX2 $dY2 $dZ2, or
REMARK TRANSLATE $dX1 $dY1 $dZ1
REMARK TRANSLATE $dX2 $dY2 $dZ2
Conclusions about re-orientation, rotation and translation
Using the rigid-body re-orientation, rigid-body rotation and rigid-body translation algorithms implement in R.E.D. III.4 and R.E.D. IV version June 2010 any user can check that:
- Translation does not affect charge values, and applying the rigid-body translation algorithm or the QMRA algorithm lead to identical charge values.
- Applying the rigid-body rotation algorithm or rigid-body re-orientation algorithm lead to identical charge values.
Consequently, we suggest to all users using the rigid-body re-orientation algorithm and its multiple re-orientation approach (i. e. the case -3-) defined in the first version of R.E.D.
-8- An initial P2N file is defined by two columns of atom names. The first column of atom names (or "yellow column" examplified in the P2N file below) is used in automatic RESP input generation, while the second one (or "red column" also examplified in the P2N file below) is involved in the conservation of international atom name conventions (i. e. CA atom name for alpha carbons of aminoacids, or C1' atom name for anomeric carbons of sugars, for instance) in the Tripos mol2 files generated by R.E.D. III.x. In the case of the absence of the second column of atom names in an initial P2N file, the atom names in Tripos mol2 files are automatically generated by R.E.D. III.x following another approach. In this case, the atom names of Tripos mol2 files are constituted by the chemical symbol of the chemical element and a number which is automatically incremented (as a consequence, the international atom name conventions defined for the molecule atoms are lost). The implementation of automatic RESP input generation in the R.E.D. versions I, II and III has to be described. Indeed, it is strongly related to the choice of creating the P2N file format.
General information about charge fitting
* In ESP charge derivation [in Weiner et al. force field, (1984 & 1986)], atom charge values are fitted to reproduce the MEP, and charge equivalencing of chemically equivalent atoms is performed a posteriori to the fit.
* In RESP charge derivation [in Cornell et al. force field (parm94.dat) and its successive adaptations (parm96.dat, parm98.dat and parm99.dat), and Duan et al. force field], atom charge values are fitted to reproduce the MEP in a two stage fit. The charge values are affected by the use of hyperbolic restraints, and charge equivalencing of chemically equivalent atoms is carried out during the fit.
- In 'standard' RESP charge derivation (i. e. originally published by the Kollman's group) a weak restraint (qwt = 0.0005) is used in charge derivation for all the heavy atoms during the first stage, and a stronger restraint (qwt = 0.001) is only applied to the methyl and methylene carbons during the second stage.
- In 'non-standard' RESP charge derivation, the use of the "qwt = 0.001" restraint in the second stage can be potentially extended to any type of carbon (in C=O, CH, and C=C, for instance), nitrogen, oxygen, silicon, phosphate and sulfur atoms. However, the use of such 'non-standard' RESP inputs is only recommended for expert users in test studies.
Rules followed by the R.E.D. program for automatic RESP input generation
R.E.D. automatically generates the inputs used in ESP, 'standard' and 'non-standard' RESP charge derivation for the RESP program based on the atom names found in the first column of atom names in initial P2N files by following three simple rules:
* The heavy atoms, whose the atom charge values have to be re-optimized in the second RESP stage (i. e. using the "qwt = 0.001" restraint) have to present in their atom name the "T" letter after their chemical symbol. With R.E.D. I, only 'standard' RESP inputs could be produced. Consequently, only methyl and methylene carbons had to present this "T" letter in their atom name. The other atom names had only to use the letter of their chemical symbol [i. e. "C" for the other carbons (in C=O, CH and C=C, for instance), "O" for oxygen, "H" for hydrogen etc...] in the definition of their atom name. With R.E.D. II, the extension of this rule to five other atoms (N, O, Si, P and S) has been implemented, and can be applied for the generation of 'non-standard' RESP inputs.
* The same number (positive integer) has to be added to this/these letter(s) for equivalent atoms (whatever the equivalencing procedure is, i. e. during the fit or a posteriori to the fit). A consequence to this is that non-equivalent atoms display different atom names.
* Each hydrogen linked to an heavy atom must have the same number as this atom.
Thus, an atom name belonging to the first column of atom names in a P2N file is defined as follows:
* HO1, 1H2 or O4' (often found in PDB files) are not valid names. Using the three rules reported above, a correct way could be H1, H2 or O4, respectively. I1 and Ag1 atom names are also rejected because their total number of electrons are above Z = 35.
* As previously said, equivalent atoms must have the same atom names [same letter(s) and number(s)]. The insightII molecular graphics program from Accelrys Inc. automatically renames the atoms, which have the same name to differentiate them. On the contrary, the VMD program displays the atom name labels without modifying them, and thus, is very convenient for checking the selected atom names written in P2N file format.
* When several conformations are available in a P2N file, only the atom names (defined with the rules previously defined) belonging to the first conformation are used in RESP input generation, and are applied to the other conformations.
Limitations of the R.E.D. versions I and II and creation of the P2N file format
With the R.E.D. versions I and II, only a single PDB file with the regular column of atom names could be used as input for a R.E.D. execution. The user had to manually modify these atom names for RESP input generation before running R.E.D. according to the rules previously defined. Although quite simple and flexible to use, such a strategy presented two important limitations:
* Such PDB atom name editing is tedious, time-consuming, and error-prone for large and multiple molecules.
* The international atom name conventions defined for those atom names are lost. This is particularly problematic when the atom names with those conventions are required in AMBER and CHARMM force field libraries.
Thus, to solve these problems, PDB files with two different types of atom names (one used in RESP input generation, and the other used to keep those atom name conventions), are now used by R.E.D. III.x as inputs. This type of PDB file has been named P2N for PDB file with two atom names (by analogy to the PQR file format).
Justification of these atom naming rules
A different strategy could have been followed for RESP input generation in R.E.D. Indeed, an algorithm based on the detection of chemical group topology could have been used for a full automatic RESP input generation. Such an approach presents the advantage that RESP inputs are automatically generated without the need of modifying PDB or P2N atom names. Thus, this approach is clearly simpler in particular for novice users. However, in our opinion it presents also strong limitations. In this case, the corresponding program is used as a "black box", meaning that the user generates charge values without understanding the scientific basis behind it. Moreover, it is rigid and can only generate a single set of RESP inputs and atom charge values for a molecule.
The R.E.D. program is a tool designed for automatic charge derivation, but also for the study and improvement of atom charge models. Thus, we decided to developed an algorithm, which allows automatic charge derivation, but lets also human beings controlling and modifying it. In our opinions, the creation of the P2N file format associated with simple rules of atom naming is a good compromise between some automaticity required in charge derivation and force field library development, and some hability of studying and improving atom charge models. Thus, by modifying atom names in P2N files one can generate many different sets of charge values based on ESP, standard and non-standard RESP charge derivation. One can also fully control charge equivalencing of chemical equivalent atoms, and group of atoms that one considers equivalent. Indeed, so far no algorithm is available allowing a general, efficient and flexible charge value equivalencing approach for groups of atoms. In these conditions, we do believe that only a human being can make the choice of making equivalent or not equivalent groups of atoms.
-9- A P2N might contain additional information about charge constraints required during a charge fitting. These constraints are intra-molecular charge constraint(s) within a molecule, inter-molecular charge constraint(s), and/or inter-molecular charge equivalencing between different molecules. Such constraints are required in charge derivation of molecule fragments and force field topology databases.
-10- Below, is an example of P2N file (or PDB file with two columns of atom names) with different comments:
REMARK TITLE Dimethylalanine-dipeptide Molecule Title REMARK CHARGE-VALUE 0 Molecule total charge value REMARK MULTIPLICITY-VALUE 1 Spin multiplicity value REMARK REMARK Conformation close to that found in an alpha helix Just a comment (not used by R.E.D.) ATOM 1 CT1 ACE 1 3.164 -0.942 -0.371 C1 ATOM 2 H1 ACE 1 3.181 -1.008 -1.453 H11 Yellow colum: First column of atom names ATOM 3 H1 ACE 1 3.877 -0.182 -0.067 H12 Required in RESP input generation ATOM 4 H1 ACE 1 3.466 -1.887 0.057 H13 (based on three simple rules) ATOM 5 C2 ACE 1 1.794 -0.579 0.161 C ATOM 6 O3 ACE 1 1.357 -1.045 1.175 O ATOM 7 N4 AIB 2 1.091 0.304 -0.605 N ATOM 8 H4 AIB 2 1.583 0.729 -1.358 H ATOM 9 C5 AIB 2 -0.094 1.027 -0.132 CA ATOM 10 CT6 AIB 2 -0.554 1.933 -1.285 CB1 Red colum: Second column of atom names ATOM 11 H6 AIB 2 0.211 2.671 -1.513 HB11 Conservation of international atom name conventions ATOM 12 H6 AIB 2 -0.759 1.355 -2.180 HB12 ATOM 13 H6 AIB 2 -1.456 2.459 -1.001 HB13 ATOM 14 CT6 AIB 2 0.222 1.874 1.106 CB2 ATOM 15 H6 AIB 2 -0.651 2.433 1.409 HB21 ATOM 16 H6 AIB 2 0.531 1.251 1.933 HB22 ATOM 17 H6 AIB 2 1.023 2.569 0.870 HB23 ATOM 18 C8 AIB 2 -1.269 0.078 0.172 C ATOM 19 O9 AIB 2 -2.127 0.412 0.943 O ATOM 20 N10 NME 3 -1.346 -1.051 -0.562 N ATOM 21 H10 NME 3 -0.518 -1.359 -1.013 H ATOM 22 CT11 NME 3 -2.369 -2.036 -0.282 C2 ATOM 23 H11 NME 3 -2.203 -2.533 0.668 H21 ATOM 24 H11 NME 3 -3.337 -1.557 -0.255 H22 ATOM 25 H11 NME 3 -2.365 -2.775 -1.074 H23 CONECT 1 2 3 4 5 CONECT 2 1 CONECT 3 1 CONECT 4 1 Atom connectivities required in Tripos mol2 file format generation CONECT 5 1 6 7 CONECT 6 5 Atom connectivities are common for different conformations available in a P2N file CONECT 7 5 8 9 => In a P2N file, all conformations of a molecule should preserve the same atom order CONECT 8 7 CONECT 9 7 10 14 18 CONECT 10 9 11 12 13 CONECT 11 10 CONECT 12 10 CONECT 13 10 CONECT 14 9 15 16 17 CONECT 15 14 CONECT 16 14 CONECT 17 14 CONECT 18 9 19 20 CONECT 19 18 CONECT 20 18 21 22 CONECT 21 20 CONECT 22 20 23 24 25 CONECT 23 22 CONECT 24 22 CONECT 25 22 TER TER character required to differentiate sets of Cartesian coordinates belonging to different conformations REMARK Conformation close to that found in an extended form Just a comment (not used by R.E.D.) ATOM 1 CT1 ACE 1 -3.285 1.356 0.006 C1 ATOM 2 H1 ACE 1 -2.800 2.297 -0.225 H11 ATOM 3 H1 ACE 1 -4.081 1.175 -0.704 H12 ATOM 4 H1 ACE 1 -3.730 1.426 0.993 H13 ATOM 5 C2 ACE 1 -2.342 0.170 -0.012 C ATOM 6 O3 ACE 1 -2.768 -0.957 -0.006 O ATOM 7 N4 AIB 2 -1.031 0.481 -0.015 N ATOM 8 H4 AIB 2 -0.752 1.437 -0.019 H ATOM 9 C5 AIB 2 0.070 -0.470 0.001 CA ATOM 10 CT6 AIB 2 0.040 -1.352 -1.260 CB1 ATOM 11 H6 AIB 2 -0.895 -1.891 -1.299 HB11 ATOM 12 H6 AIB 2 0.128 -0.741 -2.152 HB12 ATOM 13 H6 AIB 2 0.849 -2.075 -1.261 HB13 ATOM 14 CT6 AIB 2 0.031 -1.324 1.281 CB2 ATOM 15 H6 AIB 2 0.841 -2.045 1.305 HB21 ATOM 16 H6 AIB 2 0.110 -0.692 2.159 HB22 ATOM 17 H6 AIB 2 -0.904 -1.863 1.323 HB23 ATOM 18 C8 AIB 2 1.343 0.401 -0.003 C ATOM 19 O9 AIB 2 1.289 1.605 -0.011 O ATOM 20 N10 NME 3 2.518 -0.251 0.004 N ATOM 21 H10 NME 3 2.534 -1.242 0.009 H ATOM 22 CT11 NME 3 3.777 0.467 0.002 C2 ATOM 23 H11 NME 3 3.867 1.086 -0.881 H21 ATOM 24 H11 NME 3 3.861 1.101 0.875 H22 ATOM 25 H11 NME 3 4.582 -0.255 0.011 H23 END END character not required (not used by R.E.D.)
-11- The implementation of geometrical constraint(s) during geometry optimization is also available. However, in the present version this feature is limited to the interface of the Gaussian program. Four types of constraints are handled in the P2N file format by using the following formats:
- Cartesian coordinates can be constrained:
REMARK GEOM-OPT-CONSTRAINT W
- Bonds can be constrained:
REMARK GEOM-OPT-CONSTRAINT W X dst.val
- Angles can be constrained:
REMARK GEOM-OPT-CONSTRAINT W X Y agl.val
- Dihedral angles can be constrained:
REMARK GEOM-OPT-CONSTRAINT W X Y Z dhl.val
"W", "X", "Y" and "Z" stand for the atom numbers in the atom order of the considered molecule. "dst.val", "agl.val" and "dhl.val" are the distance value between the two considered atoms in Angstroms Å (0 < dst.val = 10), the angle value (0 < value = 180) between the three considered atoms in degrees and the dihedral value between the four considered atoms in degrees (positive and negative values are handled), respectively. Providing the exact constraint value(s) for a bond, an angle or a dihedral in the input geometry is strongly recommended.
Examples:
REMARK GEOM-OPT-CONSTRAINT 1
The Cartesian coordinates of the first atom in the atom order are frozen.
REMARK GEOM-OPT-CONSTRAINT 1 2 1.50
The distance between the atoms 1 and 2 is set/frozen to 1.50 Å.
REMARK GEOM-OPT-CONSTRAINT 1 2 5 6 180
The dihedral angle between the atoms 1 2 5 6 is set/frozen to 180 degrees.
Different types of constraint can be associated in a given P2N file/Gaussian input file.
-I.2.2- The Ante_R.E.D. program
Preparation of $n 'Mol_red$n.p2n' input file(s) (representing $n molecules involved in a $n molecule, $i conformation and $j orientation RESP or ESP fit) for R.E.D. III.x is a time-consuming and error-prone work. To limit this, the Ante_R.E.D. program has been developed. Ante_R.E.D. is a new PERL script provided with R.E.D. III.x. Its main objective is to transform a PDB file (generated using a molecular graphics program for instance) into a P2N file. In fact, each time Ante_R.E.D. is executed, a single PDB file is used as input and five different output files are generated (P2N, PDB file with a new atom order, GAMESS input for geometry optimization, Gaussian input for geometry optimization, and a summary of the atom connectivities detected for the molecule). Ante_R.E.D. is executed as it follows:
perl Ante_RED.pl File.pdb > Ante_RED.log
Using the PDB input file, Ante_R.E.D. automatically executes the following tasks:
- Atom re-ordering: Many graphic interfaces generate PDB files having their hydrogen atoms located in the latest positions in the atom order of a residue. This is really inconvenient in RESP charge derivation where the methyl and methylene groups have to be well identified. Thus, Ante_R.E.D. re-orders atoms in each PDB file in such a way that the hydrogen atoms are located just after the heavy atoms they are bound to.
- Default molecule title (REMARK TITLE MOLECULE), total charge value = 0 (REMARK CHARGE-VALUE 0) and spin multiplicity = 1 (REMARK MULTIPLICITY-VALUE 1) are automatically printed in the P2N file. These pieces of information have to be double checked and obviously corrected (if the total charge and spin multiplicity values generated by Ante_R.E.D. for the studied molecule are wrong). Indeed, Ante_R.E.D. does not calculate the spin multiplicity or the total charge value of a molecule.
- Based on the new atom order generated, the atom connectivities are automatically calculated and printed in the P2N file in the PDB format. However, each user should always check these connectivities in particular when the studied molecule contains a metal or when Cartesian coordinates available in the PDB file (as input to Ante_R.E.D.) are not optimized using a molecular mechanics, semi-empirical or ab initio method. A file summarizing the connectivities is also produced to describe but also detect potential errors in those connectivities.
- The original PDB atom names (atom name column "1") are moved to a new column (2nd column of atom names, or "red column" already examplified in the P2N file above) located after the "Z" Cartesian coordinates to keep the atom name conventions defined in graphic interfaces, and a new set of PDB atom names (in the atom name column "1", or "yellow column" also examplified in the P2N file above) required in the production of RESP inputs are automatically generated. In the way automatic RESP input generation has been implemented in the R.E.D. program, the atoms involved in chemically equivalent groups of atoms must have the same atom names. However, since these groups of atoms are not detected by Ante_R.E.D. (only atoms within a group of atoms are set equivalent by Ante_R.E.D.) the user must always double check the PDB atom names available in the atom name column "1". Indeed, only a user can make the decision which chemical groups are to be set equivalent, and which ones are not. See the three simple rules followed by R.E.D. to generate the inputs for the RESP program; these rules were previously defined in this tutorial.
- GAMESS and Gaussian inputs (with the new atom order) needed in the geometry optimization step are also generated by Ante_R.E.D. It is important to underline that the geometry optimization step can be directly performed within a R.E.D. execution (selecting the $OPT_Calc = "On" & $MEPCHR_Calc = "On" variables in the R.E.D. source code; this was implemented in R.E.D. II and was not modified in R.E.D. III.x). However, in a charge derivation procedure involving $n molecules * $i conformations, it is unlikely that all the geometry optimization jobs converge. Consequently, the R.E.D. run is automatically stopped when a geometry optimization job do not reach convergence. This leads to a 'FAILED' R.E.D. execution. A way to avoid this problem is to provide the geometry optimization outputs of the $n molecules as inputs to R.E.D. (selecting the $OPT_Calc = "Off" & $MEPCHR_Calc = "On" variables in the R.E.D. source code). File names of R.E.D. inputs are very specific. For each P2N file ('Mol_red$n.p2n' file name), the corresponding geometry optimization output generated by GAMESS or Gaussian ('Mol_red$n.log' file name) has to be provided by the user (the format of the geometry optimization output(s) being automatically recognized by R.E.D.). If a 'Mol_red$n.p2n' file contains the Cartesian coordinates of $i conformations, the corresponding 'Mol_red$n.log' file has also to contain the corresponding $i optimization outputs concatenated one after the others. Thus, Ante_R.E.D. generates geometry optimization inputs for QM program to allow the standalone execution of the QM geometry optimization jobs (i. e. without the need of R.E.D.). These jobs can be manually executed (and repeated) by the user until the $n * $i optimized sets of Cartesian coordinates are obtained.
-I.3- The tutorial itself
After providing general information about the R.E.D. III.x inputs above, we present the following tutorial.
-I.3.1- Preparation of the R.E.D. III.x inputs
-1- We start this tutorial by constructing the PDB files of two conformations of Dimethylalanine dipeptide using a molecular graphics program. Conformations close to an alpha helix, AIB-ConfA, and to an extended conformation, AIB-ConfE, are built. The atom order has to be identical in these two conformations. As mentioned above, no atom connectivities are available in those files, and the hydrogen atoms are located at the end of each residue. This molecule is constituted of three residues ACE-AIB-NME (N-Acetyl-dimethylalanine-N'-methylamide), and a single column of atom names is available.
In order to be able to generate charge values for the (+)NH3-terminal and (-)OOC-terminal fragments of Dimethylalanine dipeptide (see the section -I.3.2.4- of this tutorial), PDB files for Methylammonium, (+)NH3Me, and for Acetate, MeCOO(-), are also required. They are also constructed in a molecular graphics program (LEaP is quite convenient for building simple molecules).
-2- Next, the Ante_R.E.D. program is executed using those four PDB input files and the following "0Multi_Ante-RED.csh" script:
chmod +x 0Multi-Ante_RED.csh
./0Multi-Ante_RED.csh
#!/bin/csh
#
mkdir 1-Ante-RED
cp Ante_RED.pl AIB-Conf*.pdb Methylammonimum.pdb Acetate.pdb 1-Ante-RED
cd 1-Ante-RED
touch Ante_RED.log
# Ante_RED.pl is executed four times
foreach PDBFILE (*.pdb)
perl Ante_RED.pl $PDBFILE >> Ante_RED.log
end
rm Ante_RED.pl
cd ..
echo " Ante_R.E.D. execution done... "
For each input PDB file, five files are generated by Ante_R.E.D.:
| | Alpha helix conf. | Extended conf. | ||
| -A- | P2N output | P2N output | P2N output | P2N output |
| -B- | PDB output | PDB output | PDB output | PDB output |
| -C- | GAMESS input file | GAMESS input file | GAMESS input file | GAMESS input file |
| -D- | Gaussian input file | Gaussian input file | Gaussian input file | Gaussian input file |
| -E- | Atom connectivities | Atom connectivities | Atom connectivities | Atom connectivities |
As mentioned above, the atom order is identical in the two conformations of Dimethylalanine dipeptide. This is a requirement in multi-conformation RESP or ESP charge derivation using the R.E.D. program (versions II and III) !
-3- The next step is to execute four geometry optimizations using Gaussian inputs generated by Ante_R.E.D. and the following "0Multi-QM-Gaussian.csh" script (the total charge values of Methylammonium and Acetate have to be modified in the corresponding inputs):
chmod +x 0Multi-QM-Gaussian.csh 0Multi-QM-GAMESS.csh
./0Multi-QM-Gaussian.csh
#!/bin/csh
#
mkdir 2-Geom-Opt
cp 1-Ante-RED/*.com 2-Geom-Opt
cd 2-Geom-Opt
g98 < Acetate.com > Acetate-Gaussian.log
g98 < Methylammonium.com > Methylammonium-Gaussian.log
g98 < AIB-ConfA.com > AIB-ConfA-Gaussian.log
g98 < AIB-ConfE.com > AIB-ConfE-Gaussian.log
cd ..
echo " Gaussian QM geometry optimization jobs done... "
Two geometry optimizations using GAMESS inputs generated by Ante_R.E.D. and the following "0Multi-QM-GAMESS.csh" script are also executed. They will allow us comparing results obtained using the Gaussian and GAMESS programs later on in this tutorial.
./0Multi-QM-GAMESS.csh
#!/bin/csh
#
mkdir 2-Geom-Opt
cp 1-Ante-RED/*.inp 2-Geom-Opt
cd 2-Geom-Opt
rungms Acetate 00 1 > Acetate-GAMESS.log
rungms Methylammonium 00 1 > Methylammonium-GAMESS.log
rungms AIB-ConfA 00 1 > AIB-ConfA-GAMESS.log
rungms AIB-ConfE 00 1 > AIB-ConfE-GAMESS.log
cd ..
echo " GAMESS QM geometry optimization jobs done... "
Once completed, let us look at those QM outputs. In the Gaussian inputs, it is requested to compute the frequencies once the stationary point is found. Thus, one can check whether the optimized geometry of the two conformations of Dimethylalanine dipeptide and those of Acetate and Methylammonium are true minima or transition state structures. It is always a good idea to look for imaginary frequencies in a frequency output (this is a common problem when partial geometry optimization is carried out with geometric constraints). This can be done using the following UNIX command:
[fyd@lynx 2-Geom-Opt]$ grep "Frequencies" AIB-Conf*-Gaussian.log
The following output is obtained:
AIB-ConfA-Gaussian.log: Frequencies -- 51.7871 72.1379 85.3795 No imaginary frequency (i. e. no negative value)
AIB-ConfA-Gaussian.log: Frequencies -- 87.0265 107.0601 158.8691 => The optimized geometry is a true minimum
AIB-ConfA-Gaussian.log: Frequencies -- 184.7221 220.0506 255.0754
AIB-ConfA-Gaussian.log: Frequencies -- 286.6046 307.3547 334.8932
AIB-ConfA-Gaussian.log: Frequencies -- 351.9657 375.6893 403.8493
AIB-ConfA-Gaussian.log: Frequencies -- 445.9491 499.4537 558.3956
AIB-ConfA-Gaussian.log: Frequencies -- 571.9381 632.7098 683.3055
AIB-ConfA-Gaussian.log: Frequencies -- 731.9839 847.1281 899.6968
AIB-ConfA-Gaussian.log: Frequencies -- 985.2520 1000.3451 1036.0621
AIB-ConfA-Gaussian.log: Frequencies -- 1073.1434 1132.3716 1138.5566
AIB-ConfA-Gaussian.log: Frequencies -- 1156.7783 1169.1384 1255.8362
AIB-ConfA-Gaussian.log: Frequencies -- 1297.7729 1313.8954 1325.0612
AIB-ConfA-Gaussian.log: Frequencies -- 1361.0921 1396.4315 1437.6925
AIB-ConfA-Gaussian.log: Frequencies -- 1550.2670 1554.3031 1571.5044
AIB-ConfA-Gaussian.log: Frequencies -- 1603.9662 1612.1492 1628.8526
AIB-ConfA-Gaussian.log: Frequencies -- 1629.8653 1634.2643 1637.1150
AIB-ConfA-Gaussian.log: Frequencies -- 1644.6052 1654.3791 1660.0249
AIB-ConfA-Gaussian.log: Frequencies -- 1694.8444 1712.1478 1950.9241
AIB-ConfA-Gaussian.log: Frequencies -- 1972.9238 3206.3920 3216.3339
AIB-ConfA-Gaussian.log: Frequencies -- 3218.1270 3222.0035 3272.7006
AIB-ConfA-Gaussian.log: Frequencies -- 3277.1184 3287.0332 3303.5640
AIB-ConfA-Gaussian.log: Frequencies -- 3311.0476 3328.6616 3332.3635
AIB-ConfA-Gaussian.log: Frequencies -- 3339.8909 3860.4900 3918.2419
AIB-ConfE-Gaussian.log: Frequencies -- 22.1407 45.5826 57.7892 No imaginary frequency
AIB-ConfE-Gaussian.log: Frequencies -- 103.9544 107.6537 136.7960 => The optimized geometry is a true minimum
AIB-ConfE-Gaussian.log: Frequencies -- 147.7681 246.9502 254.3399
AIB-ConfE-Gaussian.log: Frequencies -- 288.8552 298.1426 318.2315
AIB-ConfE-Gaussian.log: Frequencies -- 337.5842 363.9564 423.3370
AIB-ConfE-Gaussian.log: Frequencies -- 428.7534 473.8661 558.5719
AIB-ConfE-Gaussian.log: Frequencies -- 596.6570 615.0255 709.3162
AIB-ConfE-Gaussian.log: Frequencies -- 756.7178 850.8131 872.4822
AIB-ConfE-Gaussian.log: Frequencies -- 956.1323 1028.6164 1066.5339
AIB-ConfE-Gaussian.log: Frequencies -- 1086.7833 1134.7400 1135.6776
AIB-ConfE-Gaussian.log: Frequencies -- 1159.8176 1172.6617 1264.8999
AIB-ConfE-Gaussian.log: Frequencies -- 1302.6186 1303.4472 1327.8368
AIB-ConfE-Gaussian.log: Frequencies -- 1371.8642 1405.8341 1454.7342
AIB-ConfE-Gaussian.log: Frequencies -- 1553.0698 1557.0705 1577.1962
AIB-ConfE-Gaussian.log: Frequencies -- 1610.1387 1615.9981 1621.9965
AIB-ConfE-Gaussian.log: Frequencies -- 1629.9165 1631.1573 1638.0918
AIB-ConfE-Gaussian.log: Frequencies -- 1651.9930 1658.2823 1664.7277
AIB-ConfE-Gaussian.log: Frequencies -- 1699.9748 1732.5246 1922.7654
AIB-ConfE-Gaussian.log: Frequencies -- 1939.8733 3215.4418 3221.6107
AIB-ConfE-Gaussian.log: Frequencies -- 3224.9001 3233.6174 3274.2581
AIB-ConfE-Gaussian.log: Frequencies -- 3279.3550 3292.4737 3302.4096
AIB-ConfE-Gaussian.log: Frequencies -- 3313.5776 3314.7845 3334.6211
AIB-ConfE-Gaussian.log: Frequencies -- 3337.5579 3869.0299 3925.7179
In our calculations the frequencies of Acetate and Methylammonium are all positive pointing that true minima are also achieved (data not shown).
-4-The last step consists of checking and preparing the P2N and QM output files for the R.E.D. III.x execution:
A single P2N file ('Mol_red$n.pdn') and a single QM output ('Mol_red$n.log') per molecule have to be used as inputs in a R.E.D. III.x execution, independently of the number of conformations used in the charge derivation:
In a single-conformation RESP fit, each P2N file generated by Ante_R.E.D. and each QM output produced by either GAMESS or Gaussian for the two conformations of Dimethylalanine dipeptide can be used more or less directly as inputs of R.E.D. III.x. However, chemically equivalent groups of atoms or groups of atoms considered equivalent by the user must have the same atom names (in the "atom name column 1" or "yellow column" already examplified in the P2N file above) to allow correct RESP input generation with R.E.D. In Dimethylalanine dipeptide case, the CT7 and H7 atom names have respectively to be renamed into CT6 and H6 to make the two beta methyl groups of the AIB residue chemically equivalent during the fit. The molecule title is also updated to replace "REMARK TITLE MOLECULE" (generated by Ante_R.E.D.) by something more compatible with Dimethylalanine dipeptide [i. e. "REMARK TITLE Dimethylalanine-dipeptide"; no space character is allowed in the title word], the default total charge and spin multiplicity values generated by Ante_R.E.D. have correct values. Finally, the frequency job available in the Gaussian output has to be removed from the QM output (R.E.D. II and III only recognize optimization output, and generates an error message if an optimization job followed by frequency calculations is used as input; this might be changed in R.E.D. IV).
In a multi-conformation RESP fit, the P2N files generated by Ante_R.E.D. for the two conformations of Dimethylalanine dipeptide have to be combined into a single 'Mol_red$n.p2n' file. A single set of atom connectivities is kept [the connectivities have to be carefully checked since they will be used in the topology definition of the molecule in the Tripos mol2 file format (as well as in the AMBER and CHARMM libraries)], and the molecule title is changed into "Dimethylalanine-dipeptide". The atom order is identical in the two conformations, and the two conformations are separated by the "TER" keyword. Finally, the "atom name column 1" is slightly modified: the CT7 and H7 atom names are renamed into CT6 and H6 (only required for the first conformation). The geometry optimization outputs generated by GAMESS for the two conformations of Dimethylalanine dipeptide have to be concatenated into a single file 'Mol_red1.log'. Thus, to a single P2N file always corresponds a single geometry optimization output file independently of the number of conformations of a molecule. Similar treatment is carried out using the two Gaussian outputs although the frequency calculations performed by Gaussian have to be removed from each QM output, as previously described.
R.E.D. III.x inputs (Dimethylalanine dipeptide):
Single-conf. RESP fit | Single-conf. RESP fit | ||
| -A- P2N input file | Mol_red1.p2n | Mol_red1.p2n | Mol_red1.p2n |
| -B1- GAMESS output (Compressed with bzip2) | Mol_red1.log | Mol_red1.log | Mol_red1.log |
| -B2- Gaussian output (Compressed with bzip2) (Frequency job removed) | Mol_red1.log | Mol_red1.log | Mol_red1.log |
Similarly the same approach in the P2N files and geometry optimization outputs for the Acetate and Methylammonium molecules are carried out in order to prepare them as inputs to the R.E.D. III.x program. Since the total charge of Acetate and Methylammonium is different to zero, the correct charge values ("-1" and "+1", respectively) are updated in the corresponding P2N files. The molecule title of Acetate and Methylammonium are also modified, and the two oxygen atoms of Acetate (atom names "O3" and "O4" in the "atom name column 1") are renamed into an identical atom name (atom name "O3") to make these two atoms chemically equivalent during the fit. The frequency calculations carried out by Gaussian are also removed from each QM output.
R.E.D. III.x inputs (Acetate and Methylammonium):
| -A- P2N input file | Mol_red1.p2n | Mol_red1.p2n |
| -B- Gaussian output (Compressed with bzip2) (Frequency job removed) | Mol_red1.log | Mol_red1.log |
-I.3.2- Execution of R.E.D. III.x
R.E.D. III.x runs on all UNIX flavors, Windows OS and Mac OS (see the Documentation section of the R.E.D. web site).
In the "MAIN PROGRAM" section of R.E.D. III.4, the following variables can be setup:
| $XRED = "Off" | If XRED="ON", R.E.D. is executed with the X R.E.D. tcl/tk script |
| $NP = "1" | Number of processor(s)/core(s) used in parallel in QM calculations |
| $QMSOFT = "FIREFLY" | "GAMESS", "FIREFLY" or "GAUSSIAN" can be interfaced |
| "GAMESS" means GAMESS-US or WinGAMESS, "FIREFLY" means PC-GAMESS, The latest version of Gaussian detected is interfaced (g09, g03, g98 or g94). | |
| $DIR = "Data" | Directory name where the final data are stored |
| If "$DIR" is left empty the "Data-RED" directory is automatically set up | |
| $OPT_Calc = "Off" | Geometry optimization is carried out if $OPT_Calc = "ON" |
| $MEPCHR_Calc = "On" | MEP & charges are calculated if $MEPCHR_Calc = "ON" |
| $Re_Fit = "Off" | Charges are refitted & force field library rebuilt from a previous R.E.D. run if $Re_Fit = "ON" |
| $CHR_TYP = "RESP-A1" | Eight charge models, which are described below are available: RESP-A1, RESP-A2, RESP-C1, RESP-C2, ESP-A1, ESP-A2, ESP-C1 & ESP-C2 |
Used in Cornell et al. FF, 1995 MEP: B3LYP/cc-pVTZ SCRF=(IEFPCM,Solvent=Ether) Used in Duan et al. FF, 2003 | |
Used in GLYCAM 2004 FF | |
Used in some AMBER, OPLS & CHARMM FF based simulations | |
Used in Weiner et al. FF, 1984/1986 | |
Used in some OPLS & CHARMM FF based simulations | |
In this tutorial, the X R.E.D. graphic interface of R.E.D. is not used ($XRED = "Off"), a single cpu machine is used in the QM calculations ($NP = "1"), GAMESS or Gaussian 98 are interfaced by R.E.D. ($QMSOFT = "GAUSSIAN" or $QMSOFT = "GAMESS"), and the RESP charge derivation procedure originally published by the Kollman's group is selected ($CHR_TYP = "RESP-A1").
The $OPT_Calc = "Off" and $MEPCHR_Calc = "On" variables are selected meaning that two types of inputs per molecule has to be provided by the user (a P2N file and a QM geometry optimization output for each molecule). However, one could prefer to execute R.E.D. with the $OPT_Calc = "On" and $MEPCHR_Calc = "On" variables. The advantage of this is that only the P2N input file would have to be prepared by the user. However, as mentioned above, it is very unlikely that in a charge derivation procedure involving $n molecules * $i conformations the corresponding $n * $i geometry optimization jobs converge (mainly because of the optimization convergence criteria set up in the GAMESS and Gaussian inputs).
The $Re_Fit = "On" variable can also be set up to refit charges and to rebuilt force field libraries from a previous R.E.D. run (this option is not demonstrated in this tutorial; applications of the re-fitting/re-building mode can be found in the MiniHowto of the R.E.D. III.x distribution; if $Re_Fit = "On", then $OPT_Calc = "Off" & $MEPCHR_Calc = "Off", automatically).
The R.E.D. III.x program is executed as follows, the information printed by the program is redirected to a "log" file:
perl RED-vIII.4.pl > RED-vIII.log |
Below is an example of such a R.E.D. III.x "log" file which provides different information about the tasks sequentially performed by R.E.D. III.x:
- First, the R.E.D. version and Internet home page are printed:
* Welcome to R.E.D. III.x *
RESP ESP charge Derive
http://q4md-forcefieldtools.org/RED/
- The charge model selected by the user is displayed:
---------------------------
- The molecule title (single molecule in the present example), total charge and spin multiplicity of the molecules are printed:
======================== Single molecule ================================
The molecule TITLE is "Dimethylalanine-dipeptide"
The TOTAL CHARGE value of the molecule is "0"
The SPIN MULTIPLICITY value of the molecule is "1"
==========================================================================
- The QM output used as input in R.E.D. is automatically recognized:
GAMESS
- The number of conformation(s) is automatically detected:
- The two columns of atoms names (P2N files) are automatically detected:
A 2nd column of atom names is detected
This 2nd column will be used in the PDB (& Tripos) file(s)
- The reorientation procedure (QMRA, just below) is automatically detected:
No three atom based re-orientation found in the PDB file
Re-orientation will be done according to the GAMESS Algorithm!
or (RBRA, just below):
1 re-orientation(s):
5 18 19
- The QM software used in the MEP computation and selected by the user is printed:
GAMESS
- Each step carried out in the charge derivation procedure is printed one after the other:
See the file(s) "JOB2-gam_m1-1-(X).log"
The RESP-A1 charges are being derived for molecule 1 ... [ OK ]
See the "punch2_m1" file(s)
The following Tripos Mol2 file(s) has/have been created.
Mol_m1-o1.mol2
- Finally, the cpu time (wall time) used by the charge derivation procedure is calculated:
More complex R.E.D. III.x log files are printed for charge derivation involving multiple molecules, multiple conformations and/or multiple orientations.
-I.3.2.1- Single-conformation single-orientation RESP fits for new dipeptide molecules
The next step of this tutorial is to illustrate the execution of the R.E.D. III.x program in single-conformation single-orientation RESP fits by selecting two different molecular re-orientation approaches (QMRA or RBRA procedure) for each optimized conformation of Dimethylalanine dipeptide. The molecular orientation of the optimized structure generated by the QM program (GAMESS or Gaussian) can be used (QMRA procedure, single orientation used in the fit) or re-oriented $j times before the MEP computing using the Rigid-Body Re-orientation Algorithm implemented in R.E.D. [RBRA procedure involving a single orientation ($j = 1) or multiple re-orientations ($j > 1)].
In RESP charge derivation involving the RBRA procedure one has to add keywords in the 'Mol_red$n.p2n' PDB file of each conformation of Dimethylalanine dipeptide to describe the three atoms involved in the Rigid-Body Reorientation Algorithm (i. e. "REMARK REORIENT 5 18 19" in the present example). On the contrary, if no re-orientation information is provided the QMRA procedure is automatically carried out.
Using the RBRA or QMRA procedure, the GAMESS or Gaussian QM program in MEP computation, and the two conformations of Dimethylalanine dipeptide, eight RESP charge derivations will be carried out (in different directories) in eight R.E.D. III.x runs. The following "0Multi-RED-vIII.csh" script is used to sequentially execute the different R.E.D. III.x jobs:
#!/bin/csh
#
# Sequential execution of R.E.D. III.x
# R.E.D. III.x & R.E.DD.B. Tutorial
#
echo " "; echo " R.E.D. III.x & R.E.DD.B. Tutorial"; echo " ";
foreach JOB ( 3-Charges-* )
cp RED-vIII.pl $JOB
cd $JOB
echo "$JOB"; echo " ";
perl RED-vIII.4.pl > RED-vIII.log
if -e ./RED-vIII.pl rm RED-vIII.pl
cd ..
tar cvf $JOB.tar $JOB > /dev/null
bzip2 $JOB.tar
end
The description of RESP charge derivation (single-conformation single-orientation RESP fits) using the RBRA or QMRA re-orientation procedures and using the GAMESS or Gaussian program for two optimized conformations of Dimethylalanine dipeptide is presented in the following table:
| Job no | QM program (MEP computation) | Re-orientation procedure (Single-orient. single-conf.) | Conformation | Initial P2N file |
| -1- | GAMESS | QMRA | Alpha helix | Mol_red1.p2n |
| -2- | Gaussian | QMRA | Alpha helix | Mol_red1.p2n |
| -3- | GAMESS | RBRA | Alpha helix | Mol_red1.p2n |
| -4- | Gaussian | RBRA | Alpha helix | Mol_red1.p2n |
| -5- | GAMESS | QMRA | Extended | Mol_red1.p2n |
| -6- | Gaussian | QMRA | Extended | Mol_red1.p2n |
| -7- | GAMESS | RBRA | Extended | Mol_red1.p2n |
| -8- | Gaussian | RBRA | Extended | Mol_red1.p2n |
RESP input and output files generated by R.E.D. III.x, R.E.D. III.x log files, and final Tripos files representing force field library precursors are downloadable from the following table. The whole data (compressed with bzip2) corresponding to each R.E.D. III.x execution can also be downloaded. Single-conformation single-orientation (RBRA or QMRA procedure) RESP fits:
| Job no | RESP input1 | RESP input2 | RESP output2 | R.E.D. III.x log file | Tripos mol2 file | Whole data |
| -1- | input1_m1 | input2_m1 | output2_m1 | RED-vIII.log | Mol_m1-o1.mol2 | File.tar.bz2 |
| -2- | input1_m1 | input2_m1 | output2_m1 | RED-vIII.log | Mol_m1-o1.mol2 | File.tar.bz2 |
| -3- | input1_m1 | input2_m1 | output2_m1 | RED-vIII.log | Mol_m1-o1.mol2 | File.tar.bz2 |
| -4- | input1_m1 | inp ut2_m1 | output2_m1 | RED-vIII.log | Mol_m1-o1.mol2 | File.tar.bz2 |
| -5- | input1_m1 | input2_m1 | output2_m1 | RED-vIII.log | Mol_m1-o1.mol2 | File.tar.bz2 |
| -6- | input1_m1 | input2_m1 | output2_m1 | RED-vIII.log | Mol_m1-o1.mol2 | File.tar.bz2 |
| -7- | input1_m1 | input2_m1 | output2_m1 | RED-vIII.log | Mol_m1-o1.mol2 | File.tar.bz2 |
| -8- | input1_m1 | input2_m1 | output2_m1 | RED-vIII.log | Mol_m1-o1.mol2 | File.tar.bz2 |
The following table compares charge values derived using single-conformation single-orientation (RBRA or QMRA procedure) RESP fits:
| Conf. A.H. |
Conf. A.H. |
Conf. A.H. |
Conf. A.H. |
Conf. Ext. |
Conf. Ext. |
Conf. Ext. |
Conf. Ext. | ||||
| Job no | -1- | -2- | -3- | -4- | -5- | -6- | -7- | -8- | |||
| QM soft | GAMESS | Gaussian | GAMESS | Gaussian | GAMESS | Gaussian | GAMESS | Gaussian | |||
| Atom name | QMRA | QMRA | Diff. | RBRA-1 | RBRA-1 | QMRA | QMRA | Diff. | RBRA-1 | RBRA-1 | |
| C1 | -0.1489 | -0.1469 | 0.002 | -0.1651 | -0.1651 | -0.1956 | -0.2464 | 0.051 | -0.2534 | -0.2535 | |
| H11 | 0.0443 | 0.0438 | 0.0479 | 0.0479 | 0.0596 | 0.0753 | 0.0773 | 0.0773 | |||
| H12 | 0.0443 | 0.0438 | 0.0479 | 0.0479 | 0.0596 | 0.0753 | 0.0773 | 0.0773 | |||
| H13 | 0.0443 | 0.0438 | 0.0479 | 0.0479 | 0.0596 | 0.0753 | 0.0773 | 0.0773 | |||
| C | 0.6727 | 0.6691 | 0.004 | 0.6751 | 0.6751 | 0.6332 | 0.6144 | 0.019 | 0.6207 | 0.6207 | |
| O | -0.5610 | -0.5606 | 0.000 | -0.5611 | -0.5610 | -0.5878 | -0.5799 | 0.008 | -0.5811 | -0.5810 | |
| N | -0.5977 | -0.5916 | 0.006 | -0.5787 | -0.5788 | -0.3600 | -0.3455 | 0.015 | -0.3636 | -0.3636 | |
| H | 0.3312 | 0.3285 | 0.3256 | 0.3256 | 0.2333 | 0.2230 | 0.2309 | 0.2309 | |||
| CA | 0.0886 | 0.0909 | 0.002 | 0.0649 | 0.0649 | 0.0265 | 0.0417 | 0.015 | 0.0563 | 0.0563 | |
| CB1 | -0.2051 | -0.2069 | 0.002 | -0.1929 | -0.1929 | -0.2516 | -0.1899 | 0.062 | -0.1786 | -0.1786 | |
| HB11 | 0.0729 | 0.0730 | 0.0702 | 0.0703 | 0.0851 | 0.0664 | 0.0629 | 0.0629 | |||
| HB11 | 0.0729 | 0.0730 | 0.0702 | 0.0703 | 0.0851 | 0.0664 | 0.0629 | 0.0629 | |||
| HB11 | 0.0729 | 0.0730 | 0.0702 | 0.0703 | 0.0851 | 0.0664 | 0.0629 | 0.0629 | |||
| CB2 | -0.2051 | -0.2069 | 0.002 | -0.1929 | -0.1929 | -0.2516 | -0.1899 | 0.062 | -0.1786 | -0.1786 | |
| HB21 | 0.0729 | 0.0730 | 0.0702 | 0.0703 | 0.0851 | 0.0664 | 0.0629 | 0.0629 | |||
| HB22 | 0.0729 | 0.0730 | 0.0702 | 0.0703 | 0.0851 | 0.0664 | 0.0629 | 0.0629 | |||
| HB23 | 0.0729 | 0.0730 | 0.0702 | 0.0703 | 0.0851 | 0.0664 | 0.0629 | 0.0629 | |||
| C | 0.6862 | 0.6908 | 0.005 | 0.6956 | 0.6956 | 0.6551 | 0.6351 | 0.020 | 0.6152 | 0.6152 | |
| O | -0.5668 | -0.5689 | 0.002 | -0.5701 | -0.5701 | -0.5496 | -0.5443 | 0.005 | -0.5409 | -0.5409 | |
| N | -0.4960 | -0.4978 | 0.002 | -0.4775 | -0.4775 | -0.4950 | -0.4833 | 0.012 | -0.4752 | -0.4752 | |
| H | 0.3004 | 0.2988 | 0.2919 | 0.2919 | 0.3687 | 0.3630 | 0.3637 | 0.3637 | |||
| C2 | -0.2714 | -0.2661 | 0.005 | -0.3028 | -0.3028 | -0.5180 | -0.5601 | 0.042 | -0.5683 | -0.5683 | |
| H21 | 0.1342 | 0.1328 | 0.1409 | 0.1409 | 0.2010 | 0.2125 | 0.2146 | 0.2146 | |||
| H22 | 0.1342 | 0.1328 | 0.1409 | 0.1409 | 0.2010 | 0.2125 | 0.2146 | 0.2146 | |||
| H23 | 0.1342 | 0.1328 | 0.1409 | 0.1409 | 0.2010 | 0.2125 | 0.2146 | 0.2146 | |||
| MEP point nb | 1041 | 1047 | 1046 | 1046 | 1038 | 1031 | 1027 | 1027 | |||
| RRMS | 0.119 | 0.118 | 0.119 | 0.119 | 0.142 | 0.136 | 0.136 | 0.136 |
For single molecular conformation of Dimethylalanine dipeptide, different charge values are obtained when using the QMRA procedure and either GAMESS or Gaussian program for MEP computation. In this study, a maximum charge difference of 0.06 e is observed for the beta carbon of the extended conformation of Dimethylalanine dipeptide. On the contrary, the charge values calculated using the RBRA procedure are highly reproducible (+/- 0.0001 e) no matter which QM program is used. Moreover, starting from different sets of Cartesian coordinates, the re-orientation algorithm implemented in a QM program does not always generate a unique molecule orientation for a target minimum (data not shown). This means that even using the Gaussian program (for instance), different "Standard orientations" might be generated for an optimized geometry leading to different charge values for a molecule. Consequently, we strongly recommend the use of the Rigid-Body Reorientation Algorithm implemented in the R.E.D. program.
-I.3.2.2- Single- or multi-conformation multi-orientation RESP fits for new dipeptide molecules
Since the molecule orientation of the optimized structure used in the MEP computation affects the atom charge values, one can select $j re-orientations ($j > 1, multiple RBRA re-orientation) in the charge derivation. Thus, multiple orientation RESP fit can be carried out to decrease the charge uncertainty observed when a single molecular orientation is used in the fitting process. For each conformation of Dimethylalanine dipeptide, a multiple $j = 4 re-orientations is carried out by adding the following keywords "REMARK REORIENT 5 18 19 | 19 18 5 | 6 19 20 | 20 19 6" in the initial P2N file. Each set of three atom numbers describing a rigib-body re-orientation is separated by a pipe character. In the present case, a four re-orientation RESP fit is carried out, the choice of the atoms involved in the RBRA procedure is chosen arbitrary (only heavy atoms are used), but fully controlled.
It is also known that molecule conformation affects atom charge values. Consequently, the multi-conformation RESP charge fitting approach has been developed to derive charge values which are more general and more suitable for use in molecular dynamics. In this tutorial, the use of the two conformations of Dimethylalanine dipeptide ($i = 2 conformations) in the charge derivation is described as examples of multi- or two-conformation RESP fits.
Thus, R.E.D. III.x is executed three times to generate two sets of charge values using each conformation of Dimethylalanine dipeptide, and a third set of charges involving both conformations [as GAMESS and Gaussian leads to highly similar charge values (+/- 0.0001 e) when the RBRA procedure is applied, only calculations carried out using Gaussian are now presented]:
cd 4-Charges-A-RBRA-4
perl RED-vIII.4.pl > RED-vIII.log
cd 4-Charges-E-RBRA-4
perl RED-vIII.4.pl > RED-vIII.log
cd 4-Charges-2conf-RBRA-4
perl RED-vIII.4.pl > RED-vIII.log
cd ..
P2N input files used by R.E.D. III.x, RESP input and output files generated by R.E.D. III.x, and final Tripos files representing force field library precursors are downloadable from the following table. The whole data (compressed with bzip2) corresponding to each R.E.D. III.x execution can also be downloaded. Single- or two-conformation four-orientation (RBRA procedure) RESP fits:
| Job no | Charge derivation | Conformation | Initial P2N file | RESP input1 | RESP input2 | RESP output2 | Tripos mol2 file | Whole data |
| -9- | Single-conf. four-orient. | Alpha helix | Mol_red1.p2n | input1_m1 | input2_m1 | output2_m1 | Mol_m1-o1.mol2 | File.tar.bz2 |
| -10- | Single-conf. four-orient. | Extended | Mol_red1.p2n | input1_m1 | input2_m1 | output2_m1 | Mol_m1-o1.mol2 | File.tar.bz2 |
| -11- | Two-conf. four-orient. | Both | Mol_red1.p2n | input1_m1 | input2_m1 | output2_m1 | Mol_m1-o1.mol2 Mol_m1-o2.mol2 |
File.tar.bz2 |
Let us look at the listing of files generated by R.E.D. III.x corresponding to the Job no "-11-", which involves $n = 1 molecule, $i = 2 conformations and $j = 4 orientations:
[fyd@lynx 4-Charges-2conf-RBRA-4]$ ls -l
| Files generated by R.E.D. III.x (Job no "-11-") | Description |
| esout_m1 | RESP output (see RESP manual) |
| espot_m1 | RESP input (MEP to the RESP format) for mol. $n = 1 File containing $i conf. * $j orient. MEP (2 * 4 files concatenated one after the others) |
| espot_m1-1-1 | MEP for mol. 1, conf. 1, orient. 1 |
| espot_m1-1-2 | MEP for mol. 1, conf. 1, orient. 2 |
| espot_m1-1-3 | MEP for mol. 1, conf. 1, orient. 3 |
| espot_m1-1-4 | MEP for mol. 1, conf. 1, orient. 4 |
| espot_m1-2-1 | MEP for mol. 1, conf. 2, orient. 1 |
| espot_m1-2-2 | MEP for mol. 1, conf. 2, orient. 2 |
| espot_m1-2-3 | MEP for mol. 1, conf. 2, orient. 3 |
| espot_m1-2-4 | MEP for mol. 1, conf. 2, orient. 4 |
| File4REDDB_m1.pdb | PDB file describing the $i conf. and $j orient. (optimized Cart. coordinates) (File required for a project submission in R.E.DD.B.) |
| input1_m1 | RESP input, stage 1 (File required for a project submission in R.E.DD.B.) |
| input2_m1 | RESP input, stage 2 (File required for a project submission in R.E.DD.B.) |
| JOB2-gau_m1-1-1.com | Gaussian input for MEP computation
$n mol. = 1, $i conf. = 1, $j orient. = 1 (optimized Cart. coordinates) |
| JOB2-gau_m1-1-1.log | Gaussian output for MEP computation
$n mol. = 1, $i conf. = 1, $j orient. = 1 (optimized Cart. coordinates) |
| JOB2-gau_m1-1-2.com | $n mol. = 1, $i conf. = 1, $j orient. = 2 |
| JOB2-gau_m1-1-2.log | $n mol. = 1, $i conf. = 1, $j orient. = 2 |
| JOB2-gau_m1-1-3.com | $n mol. = 1, $i conf. = 1, $j orient. = 3 |
| JOB2-gau_m1-1-3.log | $n mol. = 1, $i conf. = 1, $j orient. = 3 |
| JOB2-gau_m1-1-4.com | $n mol. = 1, $i conf. = 1, $j orient. = 4 |
| JOB2-gau_m1-1-4.log | $n mol. = 1, $i conf. = 1, $j orient. = 4 |
| JOB2-gau_m1-2-1.com | $n mol. = 1, $i conf. = 2, $j orient. = 1 |
| JOB2-gau_m1-2-1.log | $n mol. = 1, $i conf. = 2, $j orient. = 1 |
| JOB2-gau_m1-2-2.com | $n mol. = 1, $i conf. = 2, $j orient. = 2 |
| JOB2-gau_m1-2-2.log | $n mol. = 1, $i conf. = 2, $j orient. = 2 |
| JOB2-gau_m1-2-3.com | $n mol. = 1, $i conf. = 2, $j orient. = 3 |
| JOB2-gau_m1-2-3.log | $n mol. = 1, $i conf. = 2, $j orient. = 3 |
| JOB2-gau_m1-2-4.com | $n mol. = 1, $i conf. = 2, $j orient. = 4 |
| JOB2-gau_m1-2-4.log | $n mol. = 1, $i conf. = 2, $j orient. = 4 |
| Mol_m1-o1.mol2 | Final output of R.E.D. & force field library precursor Tripos mol2 file (optimized Cart. coordinates) with charges ($n mol. = 1, $i conf. = 1) (File required for a project submission in R.E.DD.B.) |
| Mol_m1-o1-qmra.pdb | PDB format (optimized Cart. coordinates) for visualization $n mol. = 1, $i conf. = 1, single orient. (QMRA procedure) For information since the QMRA procedure is not used... |
| Mol_m1-o1-rbra1.pdb | PDB format (optimized Cart. coordinates) for visualization $n mol. = 1, $i conf. = 1, orient. = 1 (RBRA procedure) |
| Mol_m1-o1-rbra2.pdb | PDB format (optimized Cart. coordinates) for visualization $n mol. = 1, $i conf. = 1, orient. = 2 (RBRA procedure) |
| Mol_m1-o1-rbra3.pdb | PDB format (optimized Cart. coordinates) for visualization $n mol. = 1, $i conf. = 1, orient. = 3 (RBRA procedure) |
| Mol_m1-o1-rbra4.pdb | PDB format (optimized Cart. coordinates) for visualization $n mol. = 1, $i conf. = 1, orient. = 4 (RBRA procedure) |
| Mol_m1-o2.mol2 | Final output of R.E.D. & force field library precursor Tripos mol2 file (optimized Cart. coordinates) with charges ($n mol. = 1, $i conf. = 2) (File required for a project submission in R.E.DD.B.) |
| Mol_m1-o2-qmra.pdb | PDB format (optimized Cart. coordinates) for visualization $n mol. = 1, $i conf. = 2, single orient. (QMRA procedure) For information since the QMRA procedure is not used... |
| Mol_m1-o2-rbra1.pdb | PDB format (optimized Cart. coordinates) for visualization $n mol. = 1, $i conf. = 2, orient. = 1 (RBRA procedure) |
| Mol_m1-o2-rbra2.pdb | PDB format (optimized Cart. coordinates) for visualization $n mol. = 1, $i conf. = 2, orient. = 2 (RBRA procedure) |
| Mol_m1-o2-rbra3.pdb | PDB format (optimized Cart. coordinates) for visualization $n mol. = 1, $i conf. = 2, orient. = 3 (RBRA procedure) |
| Mol_m1-o2-rbra4.pdb | PDB format (optimized Cart. coordinates) for visualization $n mol. = 1, $i conf. = 2, orient. = 4 (RBRA procedure) |
| Mol_red1-AIB-2Conf-Gaussian.log | R.E.D. III.x input |
| Mol_red1-AIB-2Conf.p2n | R.E.D. III.x input |
| Mol_red1.log -> Mol_red1-AIB-2Conf-Gaussian.log | Symbolic link to R.E.D. III.x input (Geometry optimization output) |
| Mol_red1.p2n -> Mol_red1-AIB-2Conf.p2n | Symbolic link to R.E.D. III.x input (P2N file containing two conformations) |
| output1_m1 | RESP output with charge and RRMS values, stage 1 (see RESP manual) |
| output2_m1 | RESP output with charge and RRMS values, stage 2 |
| punch1_m1 | RESP output with charge and RRMS values, stage 1 (see RESP manual) |
| punch2_m1 | RESP output with charge and RRMS values, stage 2 |
| qout1_m1 | Charge values, stage 1 (see RESP manual) |
| qout2_m1 | Charge values, stage 2 |
| RED-vIII.log | R.E.D. III.x log file |
Another aspect developed in R.E.D. III.x, is the use of multiple independent molecules (for instance, belonging to one study or a family of molecules) in a single R.E.D. III.x run. The "W-46" project available in the RESP ESP charge DDataBase" (or R.E.DD.B.) is an example of such an approach where the charge values of ten solvent molecules are derived sequentially and independently. Similar strategies could be applied for deriving the atom charge values for a set of ligands needed in a docking study: $n (number of ligands or decoys etc...) Mol_red$n.p2n (and $n Mol_red$n.log) have to be provided as inputs to R.E.D. III.x. In the Job no "-12-" presented below, the simultaneous derivation of RESP charges for $n molecules in a single R.E.D. III.x run (instead of executing $n R.E.D. runs for $n molecules: as shown previously Job no "-9-", "-10-" and "-11-") is performed:
cd 4-Charges-All
perl RED-vIII.4.pl > RED-vIII.log
cd ..
P2N input files used by R.E.D. III.x, RESP input and output files generated by R.E.D. III.x, and final Tripos files representing force field library precursors are downloadable from the following table. The whole data (compressed with bzip2) corresponding to a single R.E.D. III.x execution can also be downloaded. A single- or two-conformation four-orientation (RBRA procedure) RESP fit is presented:
| Job no | Charge derivation | Conformation | Initial P2N file | RESP input1 | RESP input2 | RESP output2 | Tripos mol2 file | Whole data |
| -12- | Single-conf. four orient. Single-conf. four orient. Two-conf. four orient. (Independent fit) | Alpha helix Extended Both |
Mol_red1.p2n Mol_red2.p2n Mol_red3.p2n |
input1_mm | input2_mm | output2_mm | Mol_m1-o1-mm2.mol2 Mol_m2-o1-mm2.mol2 Mol_m3-o1-mm2.mol2 Mol_m3-o2-mm2.mol2 |
File.tar.bz2 |
The following table compares charge values derived using RBRA based charge derivation procedures involving single-conformation single-orientation RESP fits (values from TABLE 6), single-conformation four-orientation RESP fits, and a two-conformation four-orientation RESP fit:
| Conf. A.H. |
Conf. Ext. |
Conf. A.H. |
Conf. Ext. |
Both Conf. | |||||
| Job no | -4- | -8- | -9- | -10- | -11- | ||||
| Atom name | RBRA-1 | RBRA-1 | Diff. | RBRA-4 | RBRA-4 | Diff. | RBRA-4 | ||
| C1 | -0.1651 | -0.2535 | 0.088 | -0.1577 | -0.2320 | 0.074 | -0.3131 | ||
| H11 | 0.0479 | 0.0773 | 0.0461 | 0.0691 | 0.0899 | ||||
| H12 | 0.0479 | 0.0773 | 0.0461 | 0.0691 | 0.0899 | ||||
| H13 | 0.0479 | 0.0773 | 0.0461 | 0.0691 | 0.0899 | ||||
| C | 0.6751 | 0.6207 | 0.054 | 0.6769 | 0.6348 | 0.042 | 0.7111 | ||
| O | -0.5610 | -0.5810 | 0.020 | -0.5628 | -0.5871 | 0.024 | -0.5691 | ||
| N | -0.5788 | -0.3636 | 0.215 | -0.5988 | -0.3532 | 0.246 | -0.5711 | ||
| H | 0.3256 | 0.2309 | 0.3300 | 0.2277 | 0.2845 | ||||
| CA | 0.0649 | 0.0563 | 0.009 | 0.0923 | 0.0355 | 0.057 | 0.1295 | ||
| CB1 | -0.1929 | -0.1786 | 0.014 | -0.1820 | -0.2208 | 0.039 | -0.2050 | ||
| HB11 | 0.0703 | 0.0629 | 0.0655 | 0.0751 | 0.0687 | ||||
| HB11 | 0.0703 | 0.0629 | 0.0655 | 0.0751 | 0.0687 | ||||
| HB11 | 0.0703 | 0.0629 | 0.0655 | 0.0751 | 0.0687 | ||||
| CB2 | -0.1929 | -0.1786 | 0.014 | -0.1820 | -0.2208 | 0.039 | -0.2050 | ||
| HB21 | 0.0703 | 0.0629 | 0.0655 | 0.0751 | 0.0687 | ||||
| HB22 | 0.0703 | 0.0629 | 0.0655 | 0.0751 | 0.0687 | ||||
| HB23 | 0.0703 | 0.0629 | 0.0655 | 0.0751 | 0.0687 | ||||
| C | 0.6956 | 0.6152 | 0.080 | 0.6858 | 0.6527 | 0.033 | 0.7160 | ||
| O | -0.5701 | -0.5409 | 0.029 | -0.5708 | -0.5537 | 0.017 | -0.5649 | ||
| N | -0.4775 | -0.4752 | 0.002 | -0.4822 | -0.4828 | 0.001 | -0.5597 | ||
| H | 0.2919 | 0.3637 | 0.2966 | 0.3582 | 0.3117 | ||||
| C2 | -0.3028 | -0.5683 | 0.266 | -0.2870 | -0.5022 | 0.215 | -0.1674 | ||
| H21 | 0.1409 | 0.2146 | 0.1368 | 0.1953 | 0.1068 | ||||
| H22 | 0.1409 | 0.2146 | 0.1368 | 0.1953 | 0.1068 | ||||
| H23 | 0.1409 | 0.2146 | 0.1368 | 0.1953 | 0.1068 | ||||
| MEP point nb | 1046 | 1027 | 1046 | 1027 | 1046 1027 | ||||
| 1041 | 1026 | 1041 1026 | |||||||
| 1048 | 1049 | 1048 1049 | |||||||
| 1043 | 1026 | 1043 1026 | |||||||
| RRMS | 0.119 | 0.136 | 0.120 | 0.145 | 0.107 |
In the present examples, charge value differences observed between each single-conformation RESP fit of Dimethylalanine dipeptide (ACE-AIB-NME) are striking for two atoms: the nitrogen atom of the AIB residue [difference = 0.215 (obtained in a single-conformation single-orientation RESP fit) and 0.246 [a single-conformation four-orientation RESP fit)] and the carbon atom of the NME residue [difference = 0.266 (a single-conformation single-orientation RESP fit) and 0.215 (a single-conformation four-orientation RESP fit)]. In AMBER force field derivation, a strategy which uses two-conformations in the fitting procedure (one being close to the alpha helix conformation, and the other to the extended conformation) has been widely applied. In the case of Dimethylalanine dipeptide, <phi, psi> dihedral values of <-65.11, -30.51> and <-179.70, -179.74> were found after geometry optimization (without the use of geometric constraint) for the alpha helix and extended conformations, respectively. Comparison of charge values involving single-conformation RESP fits and the two-conformation RESP fit shows also notable charge differences (once again the charge on the carbon atom of the NME residue displays striking deviation). The achiral dimethylalanine aminoacid is known to stabilize helix type conformations in peptides. Consequently, selecting a set of charge values suitable for molecular dynamics is not simple in this case. Studying the conformational features of reference peptides by molecular dynamics using different sets of charge values (originating from single- or multi-conformation RESP fits) could help in selecting the best set of charges for the considered residue. The lower is the value of RRMS [or "ESP relative RMS (SQRT(chipot/ssvpot))" in the RESP outputs] the better should be the charge model.
-I.3.2.3- Single- or multi-conformation multi-orientation RESP fits for the central fragment of new amino-acids
So far, we described charge derivation procedures for single molecules (or "whole" molecules). The next step in this tutorial is to provide examples of RESP charge derivation for the central fragment of a new aminoacids (AA). This will be demonstrated using once again Dimethylalanine dipeptide (ACE-AIB-NME) as example. The charge values of such a fragment are generally compatible (i. e. derived with a standard method) with previously existing ones available in a force field topology database. The charge value derivation for this central fragment is carried out using a capped aminoacid (ACE-AA-NME) where intra-molecular charge constraints, that are set to a value of zero, are used for the ACE and NME residues (i. e. for the CH3CO and NHCH3 groups of atoms; see the scheme 1).
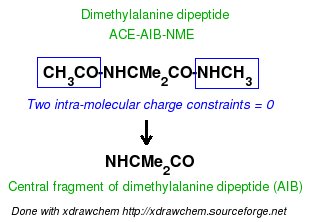
Scheme 1
In order to be handled by the R.E.D. III.x program, an intra-molecular charge constraints has to be defined for the molecule it is related to in the corresponding 'Mol_red$n.p2n' P2N file by using the "REMARK INTRA-MCC" keywords present at the beginning of a line, and followed by three fields:
(i) the value of the intra-molecular charge constraint,
(ii) the list of atom number(s) of the atom(s) involved in the intra-molecular constraint, and
(iii) a flag set to "K" or "R" (meaning that the atom(s) previously defined is/are "Kept" in or "Removed" from the Tripos mol2 file, respectively); each field is separated by the pipe "|" character.
Thus, two types of intra-molecular constraints can be used: the one maked with the "R" flag, which allows removing a part of the molecule used in the QM calculations leading to a molecular fragment, and the one marked with the "K" flag, which allows defining some compatibility with previous methods and charge values. Indeed, in the Cornell et al. force field, the four peptide bond atoms (N, H, C and O atoms) always present the same charge values for hydrophobic residues such as ALA, GLY or VAL. Consequently, these charge values are used as constraint values in the charge derivation of the central fragment of the dimethylalanine dipeptide in order to get charge values compatible with the Cornell et al. force field.
Below, are examples of inputs for setting intra-molecular charge constraints in a 'Mol_red$n.p2n' P2N file for Dimethylalanine dipeptide:
| REMARK INTRA-MCC 0.0 | 1 2 3 4 5 6 | R | Constraint value = 0 for the ACE atoms (removed from the Tripos mol2 file) |
| REMARK INTRA-MCC 0.0 | 20 21 22 23 24 25 | R | Constraint value = 0 for the NME atoms |
| REMARK INTRA-MCC -.4157 | 7 | K | Constraint value = -.4157 for the N atom of AIB (kept in the Tripos mol2 file) |
| REMARK INTRA-MCC .2719 | 8 | K | Constraint value = .2719 for the H atom of AIB |
| REMARK INTRA-MCC .5973 | 18 | K | Constraint value = .5973 for the AIB carbonyl C atom |
| REMARK INTRA-MCC -.5679 | 19 | K | Constraint value = -.5679 for the AIB carbonyl O atom |
It is important to describe the implementation of intra-molecular charge constraint(s) in R.E.D. III.x: This type of constraint is only applied in the first RESP stage, while the charge values of the atoms involved in this/these constraint(s) is/are always frozen in the second RESP stage. This is independent of the atom names defined in the 'Mol_red$n.p2n' file (in the "atom name column 1"), and in particular of the presence (or absence) of methyl or methylene groups. This method has been selected since it allows limiting the number of charge constraints in the fit. Charge constraints (intra- but also inter-molecular charge constraints) affect the RRMS value of the fit. The larger number of charge constraints the higher should be the RRMS value reflecting a decrease of the quality of the fit. Moreover, since in the charge derivation of the AIB fragment, the ACE and NME residues are removed from Dimethylalanine dipeptide, the charge values of the ACE and NME methyl groups do not need to be recomputed in the second RESP stage.
Another important aspect of charge derivation using R.E.D. III.x is that when the charge values are derived with intra-molecular constraint(s), then the corresponding calculation without constraint(s) is always automatically performed as a reference. This allows studying the impact of charge constraining on the atom charge values by comparing the two sets of charges. It is clear that if important charge differences are observed between the two sets of charges and if this corresponds to an increase of the RRMS value, the choice of the charge constraint(s) could be wrong. Generating charge values is easy; Generating good charge values using a rigorous strategy is more complex. This approach will be further developed in R.E.D. IV.
The following section demonstrates how RESP atom charges are derived for the AIB fragment (NH-CR2-CO, R = Me) using different approaches (multi-conformation multi-orientation or single-conformation multi-orientation RESP fit) realized in four executions of the R.E.D. III.x program:
-1- Single-conformation (alpha helix conformation of Dimethylalanine dipeptide) multi-orientation ($j = 4 molecular re-orientation) RESP fit using the approach named "-1-" for equivalencing charge values of atoms considered as equivalent. In this approach, the two nitrogen, hydrogen, carbon and oxygen atoms belonging to the two peptide bonds of Dimethylalanine dipeptide are considered to be equivalent. This equivalencing is imposed by providing the same atom name in the "atom name column 1" for the two carbonyl carbon atoms ("C2" atom name: "5" and "18" atom numbers), the two carbonyl oxygen atoms ("O3" atom name: "6" and "19" atom numbers), the two nitrogen atoms ("N4" atom name: "7" and "20" atom numbers), and the two hydrogen atoms ("H4" atom name" "8" and "21" atom numbers) (see the corresponding Mol_red$n.p2n file, below). This corresponds to the Job no "-13-" of this tutorial.
-2- Single-conformation (alpha helix conformation of Dimethylalanine dipeptide) multi-orientation ($j = 4) RESP fit using the approach named "-2-" for equivalencing charge values of atoms considered to be equivalent. In this approach, the two nitrogen, hydrogen, carbon and oxygen atoms belonging to the two peptide bonds of Dimethylalanine dipeptide are not considered equivalent, and consequently have different atom names in the "atom name column 1" ("C2" & "C8" atom names, "O3" & "O9" atom names, "N4" & "N10" atom names, and "H4" & H10 atom names; see the corresponding Mol_red$n.p2n file, below). This corresponds to the Job no "-14-".
-3- Single-conformation (extended conformation of Dimethylalanine dipeptide) multi-orientation ($j = 4) RESP fit (it corresponds to the Job no "-15-", and uses the equivalencing approach "-2-"),
-4- Multi-conformation ($i = 2) multi-orientation ($j = 4) RESP fit (it corresponds to the Job no "-16-", and uses the equivalencing approach "-2-").
A fifth R.E.D. execution which could correspond to a multi-molecules ($n = 2; two conformations treated as two different molecules) single-conformation multi-orientation ($j = 4) RESP fit is also carried out. This Job no "-17-" corresponds to the execution of the Job no "-14-" and "-15-" in a single R.E.D. III.x run, where each conformation is treated independently. Based on the same principle, we could have executed the Job no "-13-", "-14-", "-15-" and "-16-" in a single R.E.D. III.x run by providing the corresponding Mol_red$n.p2n and Mol_red$n.log files ($n = 1, 2, 3 and 4). A similar strategy has been already described in this tutorial, but on a "whole molecule" (see Job no "-12-").
These R.E.D. runs can be sequentially executed using a "0Multi-RED-III.csh" script similar to that used in the "-I.3.2.1-" section of this tutorial (by changing the directory names to "5-Charges-*").
Different RESP charge derivation procedures for the central fragment of Dimethylalanine dipeptide are collected in the following table. Different approaches based on single-conformation multi-orientation and multi-conformation multi-orientation RESP fits are applied:
| Job no | Charge derivation | Conformation | Initial P2N file | R.E.D. III.x log file | R.E.DD.B. project |
| -13- | Single-conf. four-orient. | Alpha helix | Mol_red1.p2n | RED-vIII.log | Not available |
| -14- | Single-conf. four-orient. | Alpha helix | Mol_red1.p2n | RED-vIII.log | F-1 |
| -15- | Single-conf. four-orient. | Extended | Mol_red1.p2n | RED-vIII.log | F-2 |
| -16- | Two-conf. four-orient. | Both | Mol_red1.p2n | RED-vIII.log | F-3 |
| -17- (-14- & -15-) | Single-conf. four-orient. (Independent fit) | Both | Mol_red1.p2n Mol_red2.p2n | RED-vIII.log | Not available |
RESP input and output files generated by R.E.D. III.x, and final Tripos files representing force field library precursors (for the whole molecule and molecule fragment) are downloadable from the following table. The whole data (compressed with bzip2) corresponding to each R.E.D. III.x execution can also be downloaded. They contain single-conformation multi-orientation or multi-conformation multi-orientation RESP fit cases:
| Job no | RESP input1 | RESP input2 | RESP output2 | Tripos mol2 file (whole molecule) | Tripos mol2 file (molecule fragment) | Whole data |
| -13- | input1_m1.sm | input2_m1.sm | output2_m1.sm | Mol_m1-o1.mol2 | Mol_m1-o1-sm.mol2 | File.tar.bz2 |
| -14- | input1_m1.sm | input2_m1.sm | output2_m1.sm | Mol_m1-o1.mol2 | Mol_m1-o1-sm.mol2 | File.tar.bz2 |
| -15- | input1_m1.sm | input2_m1.sm | output2_m1.sm | Mol_m1-o1.mol2 | Mol_m1-o1-sm.mol2 | File.tar.bz2 |
| -16- | input1_m1.sm | input2_m1.sm | output2_m1.sm | Mol_m1-o1.mol2 Mol_m1-o2.mol2 |
Mol_m1-o1-sm.mol2 Mol_m1-o2-sm.mol2 |
File.tar.bz2 |
| -17- (-14- & -15-) | input1_mm | input2_mm | output2_mm | Mol_m1-o1.mol2 Mol_m2-o1.mol2 |
Mol_m1-o1-mm2.mol2 Mol_m2-o1-mm2.mol2 |
File.tar.bz2 |
Let us look at the files generated by R.E.D. III.x for the Job no "-16-" which involves $n = 1 molecule, $i = 2 conformations and $j = 4 orientations. As previously mentioned, when a charge derivation is carried out with intra-molecular constraint(s), the analogous job without constraint is always automatically executed as a reference. Consequently, the Job no "-16-" contains the files generated without the use of intra-molecular charge constraint (as in the Job no "-11-"), as well as those that are generated using intra-molecular charge constraints. Only the files generated using the intra-molecular charge constraints and belonging to the Job no "-16-" are listed below:
[fyd@lynx 5-Charges-2conf-RBRA-4-INTRA-MCC]$ ls -l *sm*
| File generated by R.E.D. III.x (Job no"-16-") | Description |
| esout_m1.sm | RESP output (see RESP manual) Obtained with intra-molecular charge constraint (INTRA-MCC) |
| input1_m1.sm | RESP input, stage 1 Obtained with INTRA-MCC (File required for a project submission in R.E.DD.B.) |
| input2_m1.sm | RESP input, stage 2 Obtained with INTRA-MCC (File required for a project submission in R.E.DD.B.) |
| Mol_m1-o1-sm.mol2 | Final output of R.E.D. & force field library precursor Tripos mol2 file containing optimized Cartesian coordinates and charges Obtained with INTRA-MCC ($n mol. = 1, $i conf. = 1) (File required for a project submission in R.E.DD.B.) |
| Mol_m1-o2-sm.mol2 | Final output of R.E.D. & force field library precursor Tripos mol2 file containing optimized Cartesian coordinates and charges Obtained with INTRA-MCC ($n mol. = 1, $i conf. = 2) (File required for a project submission in R.E.DD.B.) |
| output1_m1.sm | RESP output with charge and RRMS values, stage 1 Obtained with INTRA-MCC |
| output2_m1.sm | RESP output with charge and RRMS values, stage 2 Obtained with INTRA-MCC |
| punch1_m1.sm | RESP output with charge and RRMS values, stage 1 Obtained with INTRA-MCC |
| punch2_m1.sm | RESP output with charge and RRMS values, stage 2 Obtained with INTRA-MCC |
| qout1_m1.sm | Charge values, stage 1 (see RESP manual) Obtained with INTRA-MCC |
| qout2_m1.sm | Charge values, stage 2 Obtained with INTRA-MCC |
Table 16 compares charge values derived using RBRA based charge derivation procedures involving single-conformation four-orientation and two-conformation four-orientation RESP fits for the whole molecule and the central fragment of Dimethylalanine dipeptide. RRMS values observed for each second RESP stage are also reported:
| Conf. A.H. |
Conf. A.H. |
Conf. A.H. |
Conf. A.H. |
Conf. A.H. |
Conf. A.H. |
Conf. Ext. |
Conf. Ext. |
Conf. Ext. |
Both Conf. |
Both Conf. |
Both Conf. | |||
| INTRA-MCC | No | Yes | Diff. | No | Yes | Diff. | No | Yes | Diff. | No | Yes | Diff. | ||
| Job no | -13- | -13- | -13- | -14- | -14- | -14- | -15- | -15- | -15- | -16- | -16- | -16- | ||
| Atom name | Charge val. | Charge val. | Charge val. | Charge val. | Charge val. | Charge val. | Charge val. | Charge val. | ||||||
| C1 | -0.3334 | -0.2838 | -0.4434 | -0.3684 | ||||||||||
| H11 | 0.0747 | 0.0629 | 0.1168 | 0.0965 | ||||||||||
| H12 | 0.1091 | 0.0963 | 0.1372 | 0.1123 | ||||||||||
| H13 | 0.1090 | 0.0946 | 0.1355 | 0.1087 | ||||||||||
| C | 0.6597 | 0.6769 | 0.6348 | 0.7111 | ||||||||||
| O | -0.5586 | -0.5628 | -0.5871 | -0.5691 | ||||||||||
| N | -0.4989 | -0.4157 | 0.083 | -0.5988 | -0.4157 | 0.183 | -0.3532 | -0.4157 | 0.063 | -0.5711 | -0.4157 | 0.155 | ||
| H | 0.2916 | 0.2719 | 0.3300 | 0.2719 | 0.2277 | 0.2719 | 0.2845 | 0.2719 | ||||||
| CA | 0.0723 | 0.1120 | 0.040 | 0.0923 | 0.1055 | 0.013 | 0.0355 | 0.1373 | 0.102 | 0.1295 | 0.1256 | 0.004 | ||
| CB1 | -0.2253 | -0.2290 | 0.004 | -0.2277 | -0.2216 | 0.006 | -0.2300 | -0.2054 | 0.025 | -0.2062 | -0.2449 | 0.039 | ||
| HB11 | 0.0785 | 0.0767 | 0.0790 | 0.0753 | 0.0766 | 0.0646 | 0.0687 | 0.0798 | ||||||
| HB12 | 0.0785 | 0.0767 | 0.0790 | 0.0753 | 0.0766 | 0.0646 | 0.0687 | 0.0798 | ||||||
| HB13 | 0.0785 | 0.0767 | 0.0790 | 0.0753 | 0.0766 | 0.0646 | 0.0687 | 0.0798 | ||||||
| CB2 | -0.2253 | -0.2290 | 0.004 | -0.2277 | -0.2216 | 0.006 | -0.2300 | -0.2054 | 0.025 | -0.2062 | -0.2449 | 0.039 | ||
| HB21 | 0.0785 | 0.0767 | 0.0790 | 0.0753 | 0.0766 | 0.0646 | 0.0687 | 0.0798 | ||||||
| HB22 | 0.0785 | 0.0767 | 0.0790 | 0.0753 | 0.0766 | 0.0646 | 0.0687 | 0.0798 | ||||||
| HB23 | 0.0785 | 0.0767 | 0.0790 | 0.0753 | 0.0766 | 0.0646 | 0.0687 | 0.0798 | ||||||
| C | 0.6597 | 0.5973 | 0.062 | 0.6858 | 0.5973 | 0.089 | 0.6527 | 0.5973 | 0.055 | 0.7160 | 0.5973 | 0.119 | ||
| O | -0.5586 | -0.5679 | 0.009 | -0.5708 | -0.5679 | 0.003 | -0.5537 | -0.5679 | 0.014 | -0.5649 | -0.5679 | 0.003 | ||
| N | -0.4989 | -0.4822 | -0.4828 | -0.5597 | ||||||||||
| H | 0.2916 | 0.2966 | 0.3582 | 0.3117 | ||||||||||
| C2 | -0.0568 | -0.1065 | -0.2504 | -0.1079 | ||||||||||
| H21 | 0.0758 | 0.0878 | 0.1270 | 0.0917 | ||||||||||
| H22 | 0.0745 | 0.0854 | 0.1272 | 0.0846 | ||||||||||
| H23 | 0.0666 | 0.0776 | 0.1182 | 0.0944 | ||||||||||
| RRMS | 0.108 | 0.148 | 0.109 | 0.140 | 0.196 | 0.156 | 0.107 | 0.136 |
As mentioned above, for each set of charge values derived for the central fragment of Dimethylalanine dipeptide (i. e. using intra-molecular charge constraints) a set of charge values for the whole molecule is generated as a reference. A maximum charge value difference between the whole molecule and the corresponding molecule fragment of 0.183 e is observed for the nitrogen atom of the AIB residue (see Table 16, alpha helix type conformation). Such comparison allows for determining the impact of intra-molecular constraints used in the charge derivation on the charge values. The increase of the RRMS values (for example from 0.108 up to 0.148 or from 0.109 up to 0.140, etc) reflects the slightly negative effect of intra-molecular charge constraints on the quality of the fit [the decrease of the RRMS value observed for the extended conformation (0.196 down to 0.156) is quite unusual, and might results from effects due to the two stage fit [indeed, the RRMS values corresponding to the first RESP stage are more understandable (data not shown, download the "File.tar.bz2" whole data file to look at the RRMS values of both RESP stages)].
Comparison of the RRMS value of the fits (0.148 or 0.140) obtained using the two different charge equivalencing procedures for the atoms involved in the two peptide bonds of Dimethylalanine dipeptide (the procedure differ by the method of how the N, H, C, and O atoms belonging to the two peptide bonds are equivalenced) also provides an argument for choosing one or the other equivalencing procedure (although the difference remains very small). This also shows that the quality of the fit usually correlates well with the number of charge constraints. The difference in the RRMS values calculated between the first and second RESP stages are also in agreement with the later statement (data not shown, download the corresponding "File.tar.bz2" whole data file to compare the RRMS values of both RESP stages).
One can derive charge values using either single conformation or two conformation RESP fit. In order to decide which of the two approaches is better the RRMS of the fit can be taken into account. In the example above the two conformation RESP fit yields lower RRMS value. One might also prefer to use charge values originating from a two conformation RESP fit since these charges are supposed to be more general and suitable for MD simulation.
-I.3.2.4- Multi-molecule multi-conformation multi-orientation RESP fits for terminal fragments of new amino-acids
-I.3.2.4.1- Implementation of inter-molecular charge constraints & inter-molecular charge equivalencing in R.E.D. III.x
Charge derivation involving a list of $n Mol_red$n.p2n molecules is a complex task. Thus, it is important to describe the implementation of multiple molecule RESP and ESP charge derivation, and the way inter-molecular charge constraint(s) and inter-molecular charge equivalencing are handled within the R.E.D. III.x program. The approach involves the two steps described below. In the first step, the charges for each of the $n molecules are derived in $n independent fits. If intra-molecular charge constraints are requested in the fit, then fitting with and without intra-molecular constraints are performed as previously described. The $n charge derivations of the $n molecules are stored in $n "Mol_$n" directories. In the second step, the charge values involving all molecules are calculated in a single fit, and stored in a "Mol_MM" (MM = multi-molecules) directory. The absence or presence of constraints between the $n molecules will affect the charge values available in this "Mol_MM" directory:
-1- In the absence of charge constraints between different molecules, the $n molecules are independent and the charges stored in each "Mol_$n" and in the "Mol_MM" directories are identical (this is under these conditions that the RESP program is recompiled with "qtol" = 0.1d-5, which ensures that charges are reproduced with the error of +/- 0.0001). This approach allows generating charge values for a defined number of independent molecules in a single fit and in a single R.E.D. III.x run. This has been already described in this tutorial (see Job no "-12-" and "-17-"), and the "W-46" project available in R.E.DD.B. is also a good example. It demonstrates how a set of molecules belonging a same family (solvent molecules) can be derived together in a single R.E.D. execution.
-2- The presence of charge constraints between the $n molecules affects the charge values obtained in the fit involving the $n molecules taken all together. Two types of charge constraints were developed within the RESP program: A) inter-molecular charge constraint and B) inter-molecular charge equivalencing. They are defined in the RESP manual, and each R.E.D. III.x user should consult this manual. The implementation of inter-molecular charge constraint(s) and inter-molecular charge equivalencing within the R.E.D. III.x program is described below. Inter-molecular charge constraint(s) and inter-molecular charge equivalencing handling within the R.E.D. III.x is limited compared to what can be done with the RESP program in a standalone execution (however, new functionalities will be added in the R.E.D. IV version):
A) Using R.E.D. III.x, an "inter-molecular charge constraint" allows setting the charge values of two groups of atoms belonging to only two different molecules to a target value (potentially an infinite number of molecules can be used when using the RESP program in a standalone way). When equal to zero, this constraint is generally useful to compute the charge values for a molecule fragment originating from the fusion between the two molecules involved in the inter-molecular constraint, the atoms used in this constraint being removed from the new structure. In this approach, one molecule (usually a small molecule) is used as a donor of a chemical group such as NH3, CO2 or PO4 for instance, while the second one (usually an aminoacid, a nucleoside or a monosaccharide) is the molecule which is going to accept this chemical group.
For instance, the use of a single inter-molecular charge constraint involving 12 atoms has been defined between the methyl group of Methylammonium, (+)NH3-Me, and the ACE-NH group of aminoacid dipeptides, ACE-AA-NME, to lead to the derivation of charge values for the (+)NH3-terminal fragment of aminoacids. A similar approach has been followed to generate charge values for the (-)OOC-terminal fragment of aminoacids involving the methyl group of Acetate, Me-COO(-), and the CO-NME group of aminoacid dipeptides. Two inter-molecular charge constraints between two molecules have also been used in the charge derivation of nucleotide fragments using dimethylphosphate and the corresponding nucleosides. In these well documented RESP charge derivation, charge fitting and the generation of the corresponding Tripos mol2 files originating from the fusion between the donor of a chemical group and the other molecule are automatically performed by R.E.D. III.x. On the other hand, the use of three or more inter-molecular charge constraints between two molecules has never been reported. Thus, charge fitting involving strictly more than two inter-molecular charge constraints are performed by R.E.D. III.x even if in some cases the charge derivation remains unknown so far. However, the corresponding Tripos mol2 files are not generated when more than two inter-molecular charge constraints are used in the charge derivation (this will be improved in the R.E.D. IV version).
An inter-molecular charge constraint is defined in one of the 'Mol_red$n.p2n' P2N files as follows: The "REMARK INTER-MCC" keywords have to be present at the beginning of a line, and are followed by four fields:
(i) the value of the inter-molecular charge constraint,
(ii) the number of the two molecules (among the $n possible) involved in the constraint: i. e. the molecule number 1 (or donor of a chemical group) and another molecule belonging to the list,
(iii) the list of atom numbers belonging to the donor of a chemical group and involved in the considered constraint, and
(iv) the list of atom numbers belonging to the second molecule, and involved in the constraint.
Each field is separated by the pipe "|" character. No flag (as in the definition of an intra-molecular charge constraint) is required since the atoms defined in an inter-molecular charge constraint are always removed from the structure generated in the Tripos mol2 format.
Below, there are examples of inter-molecular charge constraints between atoms belonging to the donor of NH3 group (molecule 1) and atoms belonging to the aminoacid dipeptide:
- A charge is constraint to the value of 0.0 between the methyl atoms of Me-NH3(+) (4 atoms) and the ACE-NH atoms (8 atoms) of Dimethylalanine dipeptide:
| REMARK INTER-MCC 0.0 | 1 2 | 1 2 3 4 | 1 2 3 4 5 6 7 8 |
- A charge is constraint to the value of 0.0 between the methyl atoms of Me-COO(-) and the CO-NME atoms of Dimethylalanine dipeptide:
| REMARK INTER-MCC 0.0 | 1 2 | 1 2 3 4 | 18 19 20 21 22 23 24 25 |
TABLE 17
An inter-molecular constraint is only applied in the first RESP stage, because the charge values of the atoms involved in such constraint are always frozen in the second RESP stage. This is independent of the atom names defined in the 'Mol_red$n.p2n' files (in the "atom name column 1"), and in particular of the presence (or absence) of methyl or methylene groups. This strategy has been already described about the implementation of intra-molecular charge constraints in R.E.D. III.x. This allows limiting the number of charge constraints in the fit, and usually leads to lower RRMS values of the fit.
B) Using R.E.D. III.x, "inter-molecular charge equivalencing" allows equivalencing the atom charge values belonging to $m molecules (where $m is the number of molecules involved in the inter-molecular charge equivalencing; the $m molecules are included in the list of $n molecules). Thus, by using inter-molecular charge equivalencing the charge values of atoms, which are not equivalent but considered equivalent by the user, can be equivalenced in the charge derivation procedure.
Inter-molecular charge equivalencing and inter-molecular charge constraint(s) are strongly coupled when used in R.E.D. III.x. Indeed, the absence or presence of inter-molecular charge constraint(s) between the molecule number 1 ('Mol_red1.p2n') and a second molecule in the list of $n molecules will pre-define inter-molecular charge equivalencing. Two cases are possible:
- The absence of inter-molecular charge constraint(s) authorizes the molecule number 1 (which is then not considered as a donor of a chemical group) to belongs to the molecules involved in inter-molecular charge equivalencing. In this case, the number of molecules involved in inter-molecular charge equivalencing, $m, is usually equal to the total number of molecules $n (or inter-molecular charge equivalencing is not applied).
- The presence of inter-molecular charge constraint(s) excludes the molecule number 1 from the group of molecules involved in inter-molecular charge equivalencing. Indeed, in this case, the molecule 1 is considered as a donor of a chemical group. Consequently, it does not make sense to involve this donor of a chemical group in inter-molecular charge equivalencing since none of its atoms can be chemically equivalent with atoms belonging to the other $m molecules. In this case, $m and $n are different ($m and $n differ by the absence or presence of 'Mol_red1.p2n', respectively).
It is sufficient to define inter-molecular charge equivalencing only in one of the 'Mol_red$n.p2n' P2N files using the "REMARK INTER-MEQA" keywords present at the beginning of a line, and followed by two fields:
(i) the numbers of the molecules involved in the constraint (if an inter-molecular charge constraint is defined in the fitting process the molecule number "1" cannot be used in inter-molecular charge equivalencing as previously said), and
(ii) the atom numbers involved in the constraints.
A strong limitation in setting inter-molecular charge equivalencing in R.E.D. III.x is that the atoms involved in inter-molecular charge equivalencing must have the same atom order (as they have the same number in molecular conformations). This will be improved in the R.E.D. IV version.
The following inputs are examples describing inter-molecular charge equivalencing:
- Charge values of atoms 1 through 8 in the molecules 1 through 4 are forced to be equivalenced:
| REMARK INTER-MEQA 1 2 3 4 | 1 2 3 4 5 6 7 8 |
- Charge values of atoms 1 through 4 in the molecules 2 through 5 are forced to be equivalenced (the molecule 1 is excluded from the list because it is considered to be a donor of a chemical group):
| REMARK INTER-MEQA 2 3 4 5 | 1 2 3 4 |
TABLE 18
Warning: in agreement with the new features related to inter-molecular charge equivalencing implemented in R.E.D. IV, the "INTER-MEQ" keyword defined for R.E.D. III.3 has been replaced by the "INTER-MEQA" one when using R.E.D. III.4. Indeed, another format for inter-molecular charge equivalencing has been defined in R.E.D. IV, and is called by using the "INTER-MEQB" keyword. "INTER-MEQB" is not recognized by R.E.D. III.4.
-I.3.2.4.2- Back to the tutorial
In order to derive RESP charges for the (+)NH3-terminal, (+)NH3CMe2CO, and (-)OOC-terminal, NHCMe2CO2(-), fragments of Dimethylalanine dipeptide the calculations need to be performed for the following three molecules: Methylammonium and Acetate, which are donors of the NH3 and CO2 chemical groups, respectively, and Dimethylalanine dipeptide (ACE-AIB-NME), which is the acceptor of a chemical group.
Below, we describe two schemes that can be applied to derive charges for the (+)NH3-terminal and (-)OOC-terminal amino acid fragments of Dimethylalanine dipeptide:
- The derivation of RESP charges for the (+)NH3-terminal fragment of Dimethylalanine dipeptide is carried out using an inter-molecular charge constraint (set to a value of 0.0) between the CH3 group of atoms of Methylammonium and the CH3CO-NH group of atoms of Dimethylalanine dipeptide, and using an intra-molecular charge constraint (set to a value of 0.0) for the NHCH3 group of atoms (in the NME residue) of Dimethylalanine dipeptide (no inter-molecular charge equivalencing is used since only two molecules are involved in the procedure; see scheme 2):
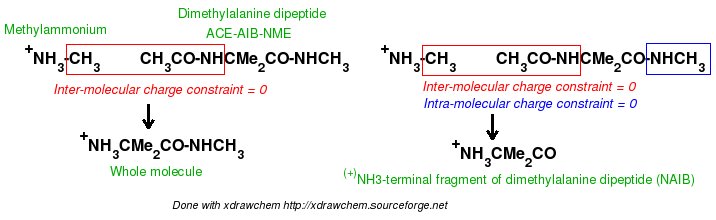
Scheme 2
- The derivation of RESP charges for the (-)OOC-terminal fragment of Dimethylalanine dipeptide is carried out using an inter-molecular charge constraint (set to a value of 0.0) between the CH3 group of atoms of Acetate and the CO-NHCH3 group of atoms of Dimethylalanine dipeptide, and using an intra-molecular charge constraint (set to a value of 0.0) for the CH3CO group of atoms (in the ACE residue) of Dimethylalanine dipeptide (no inter-molecular charge equivalencing is used since only two molecules are used in the procedure; see scheme 3):
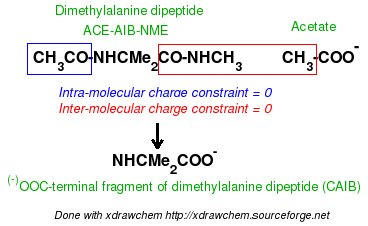
Scheme 3
The description of a multi-molecule multi-conformation and multi-orientation RESP charge derivation for the (+)NH3-terminal (Job no "-18-") and (-)OOC-terminal (Job no "-19-") fragments of Dimethylalanine dipeptide is presented in the following table. The approach involves two molecular orientations for the donor of a chemical group (Methylammonium or Acetate) and two conformations and four molecular orientations for Dimethylalanine dipeptide:
| Job no | Charge derivation | Conformation | Initial P2N file | R.E.D. III.x log file | R.E.DD.B. project |
| -18- | Mol. 1: Two-orient. Mol. 2: Two-conf. four-orient. | Single conf. Alpha helix & entended |
Mol_red1.p2n Mol_red2.p2n | RED-vIII.log | F-7 |
| -19- | Mol. 1: Two-orient. Mol. 2: Two-conf. four-orient. | Single conf. Alpha helix & entended |
Mol_red1.p2n Mol_red2.p2n | RED-vIII.log | F-11 |
RESP input and output files generated by R.E.D. III.x, and final Tripos files representing force field library precursors (whole molecule and molecule fragment) are downloadable from the following table. The whole data (compressed with bzip2) corresponding to each R.E.D. III.x execution can also be downloaded. Multi-molecule multi-conformation multi-orientation RESP fit data:
| Job no | RESP input1 | RESP input2 | RESP output2 | Tripos mol2 file (whole molecule) | Tripos mol2 file (molecule fragment) | Whole data |
| -18- | input1_mm | input2_mm | output2_mm | Mol_m1-o1.mol2 Mol_m2-o1.mol2 Mol_m2-o2.mol2 |
None available Mol_m2-o1-mm1.mol2 Mol_m2-o2-mm1.mol2 |
File.tar.bz2 |
| -19- | input1_mm | input2_mm | output2_mm | Mol_m1-o1.mol2 Mol_m2-o1.mol2 Mol_m2-o2.mol2 |
None available Mol_m2-o1-mm1.mol2 Mol_m2-o2-mm1.mol2 |
File.tar.bz2 |
Let us look at the listing of files generated by R.E.D. III.x corresponding to the Job no "-18-", which involves $n = 2 molecules ($i = 1 conformation and $j = 2 orientations for Methylammonium, and $i = 2 conformations and $j = 4 orientations for Dimethylalanine dipeptide):
[6-Charges-2conf-RBRA-4-INTER-MCC1]$ ls -d *
After running R.E.D. III.x, three directories are generated:
- The "Mol_m1" directory contains the files generated for Methylammonium which correspond to the MEP computations and to the fit involving only Methylammonium (i. e. without Dimethylalanine dipeptide). The listing of files is similar to that of the Job no "-11-" previously described (except that a single conformation and two orientations are used in the present fit; no intra-molecular charge constraint is requested in the fit).
- The "Mol_m2" directory contains the files generated for Dimethylalanine dipeptide which correspond to the MEP computations and to the fit involving only Dimethylalanine dipeptide (i. e. without Methylammonium). The listing of files is identical to that of the Job no "-16-" previously described (this case includes single intra-molecular charge constraint).
- The "Mol_MM" directory only contains the files corresponding to the charge fit involving both Methylammonium and Dimethylalanine dipeptide, the constraints [intra-molecular charge constraints, inter-molecular charge constraints and inter-molecular charge equivalencing (the latter is not used in this example)] requested for the two molecules being combined and used in the multi-molecule fit. The Tripos mol2 files corresponding to the fusion of two different molecules are also available in this directory.
The following table compares charge values derived for terminal fragments [(+)NH3-terminal (NAIB), and (-)OOC-terminal (CAIB) fragments] of Dimethylalanine dipeptide to the charge values obtained for each individual molecule (Methylammonium, Acetate, and Dimethylalanine dipeptide) used to build those molecular fragments. The fit involves a single-conformation and two-molecular orientations for the donor of a chemical group (Methylammonium or Acetate), and two-conformations and four-orientations for Dimethylalanine dipeptide. RRMS values observed for each second RESP stage are also reported:
| Job no | -18- | -18- | Job no | -19- | -19- | |
| Mol. name | MeNH3+ | NAIB fragment | Mol. name | MeCOO- | CAIB fragment | |
| Atom name | Charge val. | Charge val. | Atom name | Charge val. | Charge val. | |
| C1 | -0.0458 | ---- | C1 | -0.2100 | ---- | |
| H11 | 0.1176 | ---- | H11 | -0.0026 | ---- | |
| H12 | 0.1176 | ---- | H12 | 0.0051 | ---- | |
| H13 | 0.1192 | ---- | H13 | 0.0051 | ---- | |
| N | -0.2181 | 0.0433 | C | 0.8927 | 0.7472 | |
| H1 | 0.3032 | 0.2451 | O | -0.8451 | -0.8106 | |
| H2 | 0.3032 | 0.2451 | OXT | -0.8451 | -0.8106 | |
| H3 | 0.3032 | 0.2451 | ---- | ---- | ---- | |
| RRMS | 0.011 | ---- | RRMS | 0.008 | ---- | |
| Mol. name | AIB dipeptide | NAIB fragment | Mol. name | AIB dipeptide | CAIB fragment | |
| Atom name | Charge val. | Charge val. | Atom name | Charge val. | Charge val. | |
| C1 | -0.3684 | ---- | C1 | -0.3684 | ---- | |
| H11 | 0.0965 | ---- | H11 | 0.0965 | ---- | |
| H12 | 0.1123 | ---- | H12 | 0.1123 | ---- | |
| H13 | 0.1087 | ---- | H13 | 0.1087 | ---- | |
| C | 0.7112 | ---- | C | 0.7112 | ---- | |
| O | -0.5691 | ---- | O | -0.5691 | ---- | |
| N | -0.5712 | ---- | N | -0.5712 | -0.3821 | |
| H | 0.2845 | ---- | H | 0.2845 | 0.2681 | |
| CA | 0.1297 | 0.2223 | CA | 0.1297 | -0.0664 | |
| CB1 | -0.2063 | -0.2963 | CB1 | -0.2063 | -0.2265 | |
| HB11 | 0.0687 | 0.0913 | HB11 | 0.0687 | 0.0846 | |
| HB12 | 0.0687 | 0.0913 | HB12 | 0.0687 | 0.0846 | |
| HB13 | 0.0687 | 0.0913 | HB13 | 0.0687 | 0.0846 | |
| CB2 | -0.2063 | -0.2963 | CB2 | -0.2063 | -0.2265 | |
| HB21 | 0.0687 | 0.0913 | HB21 | 0.0687 | 0.0846 | |
| HB22 | 0.0687 | 0.0913 | HB22 | 0.0687 | 0.0846 | |
| HB23 | 0.0687 | 0.0913 | HB23 | 0.0687 | 0.0846 | |
| C | 0.7160 | 0.6163 | C | 0.7160 | ---- | |
| O | -0.5648 | -0.5722 | O | -0.5648 | ---- | |
| N | -0.5597 | ---- | N | -0.5597 | ---- | |
| H | 0.3117 | ---- | H | 0.3117 | ---- | |
| C2 | -0.1079 | ---- | C2 | -0.1079 | ---- | |
| H21 | 0.0917 | ---- | H21 | 0.0917 | ---- | |
| H22 | 0.0846 | ---- | H22 | 0.0846 | ---- | |
| H23 | 0.0944 | ---- | H23 | 0.0944 | ---- | |
| RRMS | 0.107 | 0.043 | RRMS | 0.107 | 0.042 |
The comparison of charge values derived for the Dimethylalanine dipeptide terminal fragments to the charge values obtained for the molecules used to build those fragments clearly show the impact of the intra- and inter-molecular charge constraints on the charge values (the differences are not displayed in the table, but can be easily deduced). The RRMS values (0.043 and 0.042 for N- and C-terminal fragments, respectively) obtained in each fit corresponding to the molecular fragments demonstrate the excellent quality of the fits.
-I.3.2.5- Multi-molecule multi-conformation multi-orientation RESP fits for other fragments
- This tutorial has only presented RESP charge derivation procedures for whole molecules and molecular fragments (central and charged terminal fragments) of new aminoacids by considering the Dimethylalanine dipeptide as an example. The charge values obtained are required in molecular mechanics based simulations using the Cornell et al. force field and its different adaptations [the following force field parameter files are provided in the AMBER distribution: parm94.dat (Cornell et al. force field), parm96.dat (Kollman et al. force field), parm98.dat (Cheatham et al. force field) and parm99.dat (Wang et al. force field)]. The derivation of RESP charges for neutral terminal fragments of new aminoacids might be carried out using similar strategy to the one used for the charged terminal fragments, i. e. using a donor of a chemical group [NH2 (Methylamine) and COOH (Acetic acid)] and the new aminoacid dipeptide to parameterize as acceptor of a chemical group. Projects describing charge derivation procedures of the NH2-terminal and HOOC-terminal fragments are also available in R.E.DD.B. (see the projects F-15 and F-19, respectively).
The derivation of RESP charges needed for the Duan et al. force field can be carried out by following similar approaches to those described in this tutorial. However, the theory level required to compute the MEP is different [B3LYP/cc-pVTZ theory level in diethylether: Polarized Continuum Model (PCM) using the Integral Equation Formalism (IEF)], and the source code of R.E.D. III.x has to be slightly modified. When using the Gaussian program, the Gaussian route has to be modified (see the R.E.D. III.x execution section described previously in this tutorial): the "HF/6-31G*" keyword has to be replaced by the "B3LYP/cc-pVTZ SCRF=(IEFPCM,Solvent=Ether)" ones. The IEFPCM method is also available in the GAMESS program. See also the R.E.DD.B. projects F-65 up to F-70 for charge derivation for the central fragments of unusual aminoacids derived from the cysteine residue that are compatible with Duan et al. force field.
- The derivation of RESP charges for new nucleosides and nucleotide fragments useful in modeling of the RNA and DNA was not presented in this tutorial. However, it can be easily carried out using approaches similar to those described for aminoacids. Usually, RESP charge derivation for new nucleotide fragments is performed using Dimethylphosphate (conformation gauche, gauche), as a donor of phosphate atoms and the new nucleoside. Examples of RESP charge derivation for unusual nucleotides are available in R.E.DD.B. (see for instance, the F-57, F-58 and F-59 projects). In this case, two inter-molecular charge constraints are required for the two methyl groups of Dimethylphosphate and the HO3' and HO5' hydroxyls of the target nucleoside (no intra-molecular charge constraint is needed in such an approach). In order to build proper set of nucleotides for molecular modeling the charges for four different units: the nucleoside itself, the nucleotide central fragment, and the nucleotide HO3'- and HO5'-terminal fragments (see scheme 4) need to be determined. A new tutorial is going to be written demonstrating charge derivation for unusual nucleotides.
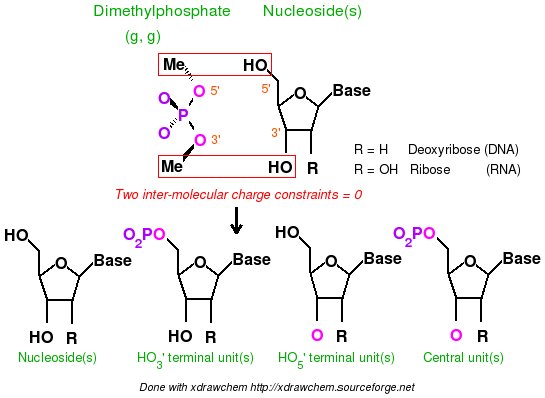
Scheme 4
In order to do this, one has to set two inter-molecular charge constraints in one of the 'Mol_red$n.p2n' R.E.D. III.x input files. Below, are examples of inter-molecular charge constraints between the two Dimethylphosphate methyl atoms and the HO3' and HO5' hydroxyl atoms of a nucleoside. The first set of constraints assigns a total charge of 0.0 to the first Dimethylphosphate methyl atoms (atom numbers 1 to 4) and the HO5' atoms (atom numbers 1 and 2) belonging to the nucleoside. A second set of constraints assigns a total charge of 0.0 to the second Dimethylphosphate methyl atoms and the nucleoside HO3' atoms:
| REMARK INTER-MCC 0.0 | 1 2 | 1 2 3 4 | 1 2 |
| REMARK INTER-MCC 0.0 | 1 2 | 10 11 12 13 | 3 4 |
TABLE 22
In order to derive RESP charge values for the whole force field topology database for DNA and/or RNA, more complex RESP charge derivation involving Dimethylphosphate and the four DNA and/or RNA nucleosides have to be performed. Such projects (16 components of a DNA force field topology database: F-45, 16 components of a RNA force field topology database: F-51, 32 components of a RNA and DNA force field topology database: F-60) are also available in R.E.DD.B. In these cases, five 'Mol_red$n.p2n' ($n = 1 to 5) files (Dimethylphosphate and the four DNA or RNA nucleosides) and nine 'Mol_red$n.p2n' ($n = 1 to 9) files (Dimethylphosphate and the eight DNA and RNA nucleosides) are used in the charge derivation procedure. Besides the inter-molecular charge constraints described in table 22, inter-molecular charge equivalencing has be to used between different nucleosides to make the charge values of the deoxyribose atoms (excluding the C1' and H1' atoms) equivalent. Inter-molecular charge equivalencing has been already described in this tutorial, but below another example is provided for illustrating inter-molecular charge equivalencing, applied to a DNA force field topology case: The charge values of the deoxyribose atoms (except the C1' and H1' atoms) i. e. the atom numbers: 1 to 4, and 7 to 17 of the four regular DNA nucleosides are made equivalent. This requires that the order of the sugar atoms in the nucleoside 'Mol_red$n.p2n' files has to be identical. The molecule number 1, Dimethylphosphate, is excluded from setting inter-molecular charge equivalencing because it is considered to be a donor of a chemical group:
| REMARK INTER-MEQA 2 3 4 5 | 1 2 3 4 7 8 9 10 11 12 13 14 15 16 17 |
TABLE 23
- The derivation of RESP charges for new monosaccharide units can be carried out using the strategies presented here in this tutorial (see scheme 5). The GLYCAM force fields are specially designed for polysaccharides, and glycoconjugates, and are distributed together with the AMBER program. To develop new fragments or units that comply with the GLYCAM force fields one also needs to derive RESP charge values. However, the authors of the GLYCAM force fields use the CHELPG algorithm (instead of the Connolly surface as in AMBER force fields) in order to compute the MEP, and a single RESP stage with a hyperbolic restraint equal to 0.01. This charge derivation approach is automatically carried out by R.E.D. III.x by selecting the $CHR_TYP = "RESP-C2" variable in the main section of the R.E.D. III.x program. To derive RESP charges for new monosaccharide fragments, one could use the hydroxyl or O-methyl group (1R chemical group bound to the C1' sugar atoms in scheme 5) with an intra-molecular charge constraint for this chemical group. However, it is important to underline that the total charge value of units in GLYCAM force fields are not always integer numbers. For instance, the total charge of the α-Galacto and β-Galacto units ("1LA" and "1LB" in the GLYCAM 2004 force field topology database, respectively) is equal to 0.194 e. This means that the intra-molecular charge constraint value used for the 1R chemical group has in this case to be a non-integer number ("X.X" total charge value in the scheme 5). Discussions about charge derivation for GLYCAM 2004 force fields can be found in the AMBER mailing list archives (i. e. there and there). A new tutorial is going to be written demonstrating charge derivation for complex oligosaccharides.
Scheme 5
-I.3.3- The Tripos mol2 file format
-I.3.3.1- Advantages of the Tripos mol2 file format
The Tripos mol2 file format has been selected as the format to create the final data generated by the R.E.D. program. In our opinion, this file format presents many advantages and is very powerful. As in the PDB format, Cartesian coordinates (very intuitive compared to the internal coordinates), atom names, atom numbers, residue names and residue numbers are available in the Tripos file format. This format has the following advantages over the PDB format:
- Many digits after the decimal point can be used to describe the Cartesian coordinates of a structure. Thus, optimized Cartesian coordinates generated by a QM program can be written to the Tripos mol2 file format without the rounding off step required for the PDB format. Consequently, recalculating an optimized structure by QM programs using data from a Tripos file is very convenient and straightforward.
- Force field atom types and atom partial charges are also available in this file format while they are absent in the PDB format (this is a strong limitation of the PDB format; this is a reason why the PQR format has been created).
- Bonds (and bond types) can also be described while they are not always present in the PDB format. Bonds are very important since they reflect the topology of the molecule. Molecular topology description is required in force field simulation, the topology is constant in different molecular orientations and conformations of a molecule observed during a simulation. Molecular topology can also be identical for two enantiomers and different diastereoisomers (if the order of atoms is the same in the stereoisomers).
Moreover, all these pieces of information are available in a single file as opposed to many other formats. Additionally, more and more programs started to recognize this file format. For instance, it is handled by Babel or Openbabel, the well known format converter programs. The recognition of this format has been incorporated in the LEaP version of AMBER 8, and this format is usually recognized by commercial packages. This file format can also be easily converted into the CML file format using a perl script to display molecular structure and charge values within Jmol (this is the strategy employed in R.E.DD.B.).
-I.3.3.2- Why do we use the Tripos mol2 file format in R.E.D. ?
One of the reason of applying the Tripos mol2 file format in the R.E.D. program is that it is force field independent. This means that the force field atom types are not generated by R.E.D. III.x, or do not need to be generated by R.E.D. III.x, and are simply replaced by chemical elements (periodic table). The charge values computed by R.E.D. can be used not only by one force field, but by any force fields (for examples by AMBER, CHARMM, OPLS and GLYCAM force fields), while specific molecular mechanics programs can be used to assign its own atom types, accordingly. For instance, the Antechamber program can be used to add General Amber force field atom types into Tripos mol2 files generated by R.E.D. III.x for small organic molecules. The Cornell et al., CHARMM or GLYCAM force field atom types for DNA, RNA and/or monosaccharide fragments can be added using script based approaches. The Openbabel program can also perform atom typing.
-I.3.3.3- Using the Tripos mol2 file format to visualize structures and charge values within an Internet Browser
Jmol associated with Java (JRE) allow visualizing within any recent Internet browsers under any operating systems (all the UNIX flavors, MAC OS and Windows) structures and charge values generated by R.E.D. This feature provides efficient and totally free graphic interface for research and teaching purposes. One has simply to convert the Tripos mol2 file generated by R.E.D. into a CML file. This can be easily carried out using the "mol2tocml" perl script provided below. The following commands can be executed in order to convert each Tripos mol2 file into the corresponding CML file:
mol2tocml.pl Your-mol2-file.mol2 or
mol2tocml.pl DIR/sub-DIR/../*.mol2
Remark: with the last versions of Jmol the Tripos mol2 file format is now fully recognized making this mol2tocml.pl script not useful anymore! Consequently, the mol2 file format is now directly used to display molecules and molecular fragments using Jmol in R.E.DD.B.
Examples of such a visualization has been automated for each project submitted in R.E.DD.B. (See for instance the W-46 and F-60 R.E.DD.B. projects).
-I.3.3.4- Using the Tripos mol2 file format as library precursor for AMBER or CHARMM force field library
Finally, let us describe how to use a Tripos mol2 file generated by R.E.D. within molecular mechanics packages and more specifically in AMBER and CHARMM. This file format is converted into an internal format, a library specific to AMBER (LEaP OFF file) or CHARMM (RTF or PSF file). The goal is to introduce additional information about the structure, which is not available or cannot be contained in the Tripos mol2 file format. Consequently, the Tripos mol2 file format can be seen and is used as a library precursor. Below we distinguish the whole molecule and molecular fragment cases.
-I.3.3.4.1- Whole molecule case
In the case of whole molecules (such as a ligand bound to DNA or a protein, or a solvent molecule), the atom types related to a specific force field have to be added to the Tripos mol2 file before converting it into a AMBER LEaP OFF or CHARMM RFT or PSF library.
- For AMBER, LEaP scripts can be used to add Cornell et al. force field atom types and convert the Tripos mol2 file format into a LEaP OFF library (the latter being the file used to generate the well known prmtop and prmcrd files). The atom types defined in the Cornell et al. force field are used in most of the AMBER and GLYCAM force fields (protein, nucleic acid and sugar force fields). Examples of such a script are available below and also in many R.E.DD.B. projects.
As an example, one can use each of the Tripos mol2 file generated by R.E.D. III.x in the Job no "-11-" of this tutorial. First, let us load in XLEaP (AMBER8 or AMBER9) the two Tripos mol2 files (containing the two conformations of Dimethylalanine dipeptide with a single set of charge values) generated by R.E.D. III.x (Figure 2A), and display the charge values (Figures 2B and 2C). Then, general information about the three units, ACE-AIB-NME, available in the Tripos mol2 files are set, and the force field atom types compatible with the Cornell et al. force field are introduced (Figure 2D, see also the LEaP script below to display the LEaP commands required to perform the operations just cited with many comments available):
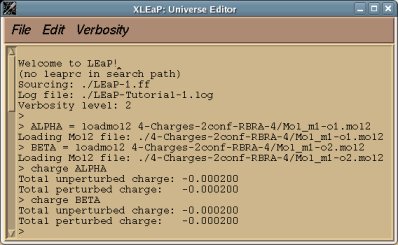
Figure 2A
 | 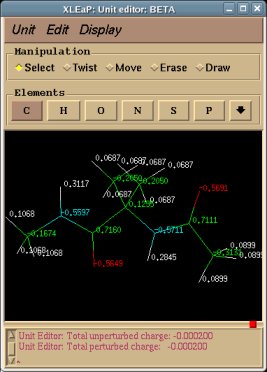 | 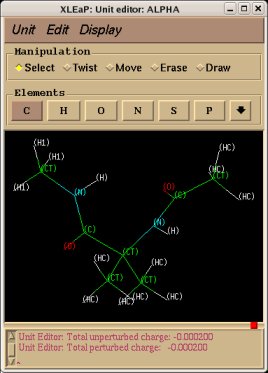 |
The whole procedure can be executed with "tLEaP" in a non-graphical mode using the LEaP-1.ff script available below. The OFF library (conformation and orientation independent) and LEaP log file are automatically generated:
tleap -f LEaP-1.ff
R.E.DD.B. can now store such a LEaP script. An example of more complex LEaP script involving ten whole molecules (solvent molecules) is available for the W-46 project.
For organic molecules, one can also use Antechamber and the following command to generate force field atom types compatible with the General Amber Force Field for the Tripos mol2 file generated by R.E.D. III.x:
antechamber -i Mol_RED-III.mol2 -fi mol2 -o Mol_GAFF.mol2 -fo mol2
- For CHARMM, it should be possible to write scripts or programs to convert the Tripos mol2 file format generated by R.E.D. III.x into a CHARMM RTF or PSF library. We are working on this and would be interested in any feedback on this. R.E.DD.B. can now store such a script for CHARMM; see for instance, the W-46 project.
-I.3.3.4.2- Molecule fragment case
For a molecule fragment (i. e. a part of a whole molecule), besides force field atom types, one has to describe the atoms in the fragment which are involved in the connections with other fragments before converting the Tripos mol2 file into AMBER OFF or CHARMM RTF or PSF library.
- For AMBER, a LEaP script can be used to add Cornell et al. force field atom types, add the description of the atoms involved in the connections with other fragments (i. e. the head and the tail), and convert the Tripos mol2 file into LEaP OFF library.
As an example, one can use the Tripos mol2 files generated by R.E.D. III.x in the Job no "-16-", Job no "-18-" and Job no "-19-" of this tutorial. First, let us load in "XLEaP" three Tripos mol2 files (only alpha helix conformation of Dimethylalanine dipeptide is used since both conformations involved in the fit lead to a single set of charge values) generated by R.E.D. III.x (Figure 3A) corresponding to the (+)NH3-terminal (NAIB unit), central (AIB unit) and (-)OOC-terminal (CAIB unit) fragments of Dimethylalanine dipeptide, and display the charge values (Figures 3B, 3C and 3D, respectively) for each of these units.
A striking feature of the NAIB and CAIB units is presented in purple in Figures 3B and 3D. Indeed, since the NH3 and COO chemical groups come from Methylammonium and Acetate, and since they are respectively fused with the CHCMe2CO and NHCHCMe2 fragments of Dimethylalanine dipeptide, the position of the NH3 and COO chemical groups in the NAIB and CAIB terminal units might look quite unusual (the Cartesian coordinates of NH3 and COO are not recomputed). However, it is important to underline that this is neither a bug nor a problem since only the topology of the unit matters when one has the objective of preparing an AMBER or CHARMM library (the Cartesian coordinates of the atoms are not that important in a library). That being said, using XLEaP (not yet available in tLEaP), the Cartesian coordinates of the NH3 and COO chemical groups can be recomputed in the NAIB and CAIB units (see Figures 3E and 3F) by first loading a force field, setting the force field atom types, and then relaxing the selected Cartesian coordinates (edit target UNIT, select atoms to relax, Edit/Relax selection).
Remark: This limitation is now solved and the geometry of a fragment originating from the fusion between two molecules is recalculated with R.E.D. III.4 and R.E.D. IV version June 2010.
As mentioned above, the atoms belonging to the NAIB, AIB and CAIB units that are involved in the connections with other units have to be declared in LEaP. Thus, one has to set the carbonyl carbon of the NAIB unit as the 'tail' atom (no 'head' is possible in NAIB since NAIB it is a N-terminal unit), the nitrogen atom of the CAIB unit as the head atom (no 'tail' is possible in CAIB since CAIB is a C-terminal unit), and the carbonyl carbon and nitrogen atoms of the AIB unit as the head and tail atoms, respectively. To set head and tail atoms for NAIB, AIB and CAIB units, the following LEaP commands can be used:
# AIB UNIT
set AIB head AIB.1.N
set AIB tail AIB.1.C
set AIB.1 connect0 AIB.1.N
set AIB.1 connect1 AIB.1.C
# NAIB UNIT: head = null
set NAIB tail NAIB.1.C
set NAIB.1 connect0 NAIB.1.N
set NAIB.1 connect1 NAIB.1.C
# CAIB UNIT: tail = null
set CAIB head CAIB.1.N
set CAIB.1 connect0 CAIB.1.N
set CAIB.1 connect1 CAIB.1.C
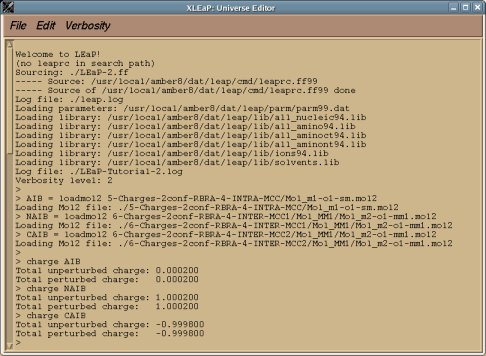
Figure 3A
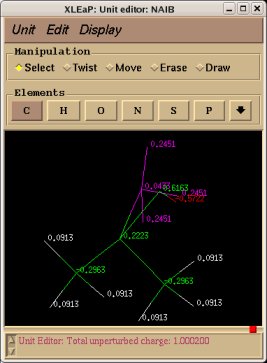 | 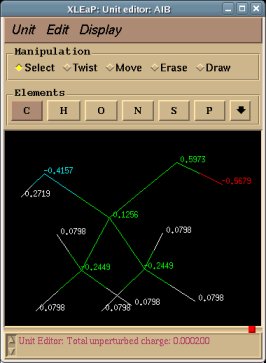 | 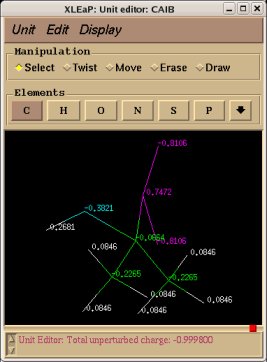 |
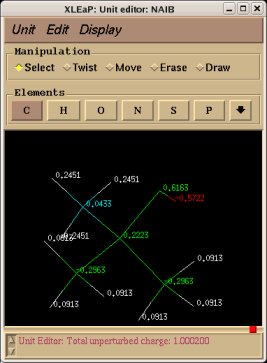 | 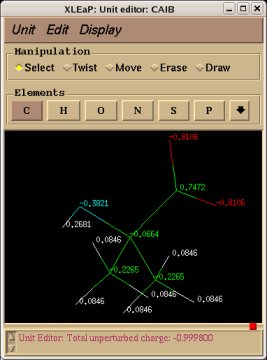 | |
As an example, one can use the NAIB, AIB and CAIB units to build the NAIB-AIB-CAIB peptide in LEaP. The "sequence" command is used to build the target peptide (Figure 3G), and the "impose" one to set the dihedral of the peptide backbone to 180.0 deg to lead to extended conformation (Figure 3H):
# NAIB-AIB-CAIB peptide
EXAMPLE = sequence { NAIB AIB CAIB }
impose EXAMPLE {1 2 3}{{N CA C N 180.0}{C N CA C 180.0}{CA C N CA 180.0}{CA C N CA 180.0}{N CA C OXT 180.0}{C CA N H1 180.0}}
saveamberparm EXAMPLE prmtop prmcrd
 |  |
Remark: The total charge of the NAIB-AIB-CAIB peptide is equal to 0.0006 e. By manually adding -0.0001 e (the charge values generated by R.E.D. have a charge uncertainty of +/- 0.0001 e) to the charge value of the nitrogen and carbonyl carbon atoms of the NAIB, AIB and CAIB units, the total charge of each unit would become an integer charge value (+1.0000, 0.0000 and -1.0000, respectively), and the total charge of NAIB-AIB-CAIB would equal zero.
The whole procedure can be executed with "tLEaP" using the LEaP-2.ff script available below. The NAIB, AIB, and CAIB OFF libraries, and the LEaP log file are automatically generated:
tleap -f LEaP-2.ff
More complex LEaP scripts are already available in R.E.DD.B. (see for instance the F-1 and F-60 projects).
- In the case of CHARMM, as for the whole molecule case, it should be possible to write scripts or programs to convert each Tripos mol2 file generated by R.E.D. III.x into a CHARMM library. This option will be available in the future versions of the R.E.D. program. R.E.DD.B. can now store such a script for CHARMM; see for instance, the F-60 project.
Should you find any mistake in this tutorial, please, send me an e-mail:
If you have questions about this tutorial, please, send your emails to the q4md-forcefieldtools mailing list. We will answer to the queries about the q4md-forcefield tools in the Amber or CCL mailing lists as well.
Last update of this tutorial page: January 12th, 2012.
Charge derivation data free for download.
Université de Picardie Jules Verne. Sanford Burnham Prebys Medical Discovery Institute.
© 2009-2024. All rights reserved.
