Introduction
Peptide nucleic acid (PNA) is a synthetic analog of DNA in which the 2-aminoethyl-glycine linkage replaces the sugar phosphodiester backbone. PNA is a non-ionic, achiral biopolymer which is not susceptible to hydrolytic cleavage. It presents key features for the development of gene therapeutic agents as it binds with high sequence selectivity to DNA and RNA and form heteroduplexes.
[1]
A set of RESP charges was previously reported for PNA,
[2] but this ensemble is not available from the publication. Moreover, no information about the molecular conformation of the PNA residues involved in charge derivation is provided, and the theory level reported in molecular electrostatic potential (MEP) computation is not compatible with the Amber additive force field model. Consequently, we report here on a new force field topology database (FFTopDB;
i.e. RESP charges embedded in a set of 24 force field libraries provided in the
Tripos mol2 and
mol3 file formats) for regular PNA with the full description of the corresponding computational conditions. This FFTopDB is compatible with the Cornell
et al. force field and its successive adaptations,
[3] and is devoted to condensed phase molecular dynamics simulations.
Computational details
In this work, the "building block" procedure applied for the Amber FFTopDB for the regular amino acid residues has been followed.
[4] Calculations were performed by using
N-acetyl-2-aminoethyl-glycine-
N'-methylamide units with purine and pyrimidine bases linked to the backbone by methylene carbonyl bonds. Six nucleobases (adenine, cytosine, guanine, hypoxanthine, thymine and uracil) were considered. The charge derivation procedure and force field library building were automatically carried out by using the R.E.D. IV program available at R.E.D. Server.
[5] This allows rigorously controlling the different parameters, which affect charge values compatible with the non-polarizable RESP charge model. Geometry optimization and MEP computation were carried out by quantum mechanical methods by using the
Gaussian 2009 (version A.01) program. The levels of theory used in geometry optimization and MEP computation were chosen to be compatible with the Cornell
et al. force field. Hence, the geometries of 36 elementary building blocks (see Figure 1a) were optimized using the HF/6-31G* level of theory.
[4] A single conformation for each PNA building block was involved in charge derivation. Geometry optimization was initiated from PNA building blocks extracted from an experimental RNA-PNA heteroduplex solved by NMR,
[6] and carried out without any geometrical constraint in gas phase.
[7] By opposition to the work of He
et al.,
[2] MEP computation employed the Connolly surface algorithm and the HF/6-31G* level of theory, thus taking into account implicit polarization required in condensed phase molecular dynamics simulations when using the Amber additive force field model.
[3] For each building block one or two pairs of molecular orientations based on the rigid-body reorientation algorithm (RBRA) implemented in the R.E.D. program were considered in MEP computation ensuring the reproducibility of the derived charge values.
[8] A total of 18 molecular fragments were generated by setting specific intra- and inter-molecular charge constraints between well-characterized connecting groups during the charge fitting step (see Figure 1b). The central fragment of each PNA residue was obtained by imposing two intra-molecular charge constraints for zeroing sum of charges on the
N-acetyl and
N'-methylamide capping groups. The
N-terminal fragment of the PNA residues was constructed by combining methylammonium and
N-acetyl-2-aminoethyl-glycine-
N'-methylamide derivatives in charge derivation. This fragment was generated by setting two charge constraints to a value of zero during the fitting step: (i) an inter-molecular charge constraint between the methylammonium methyl group and the
NH-acetyl group of each PNA unit, and (ii) an intra-molecular charge constraint for the PNA unit
N'-methylamide group. The
C-terminal fragment of the PNA residues was built following a similar approach using acetate and
N-acetyl-2-aminoethyl-glycine-
N'-methylamide derivatives. This fragment was obtained by constraining the total charge of two groups of atoms to the zero value. One group involved the acetate methyl group and the CO-
N'-methylamide group of each PNA unit, and the other one the PNA unit
N-acetyl group. As a result, the total charges of the central,
N- and
C-terminal fragments of PNA units took integer values. Force field libraries for the molecular fragments were built by removing the atoms involved in the charge constraints from the molecules involved in charge derivation, and by adding a new atom connectivity between the methylammonium nitrogen atom and the PNA C2' carbon atom for the
N-terminal fragment, and between the acetate carboxylate carbon and the PNA C5' carbon atom for the
C-terminal fragment. Inter-molecular charge equivalencing was used to force the atomic charges between the common elements of the different building blocks to be equivalent leading to a highly consistent set of charge values within the FFTopDB. In this approach, the charge values of the PNA backbone were made equivalent.
[9] RESP charge fitting was carried out by using a standalone version of the RESP program, and following the two RESP stage fitting procedure.
[10]
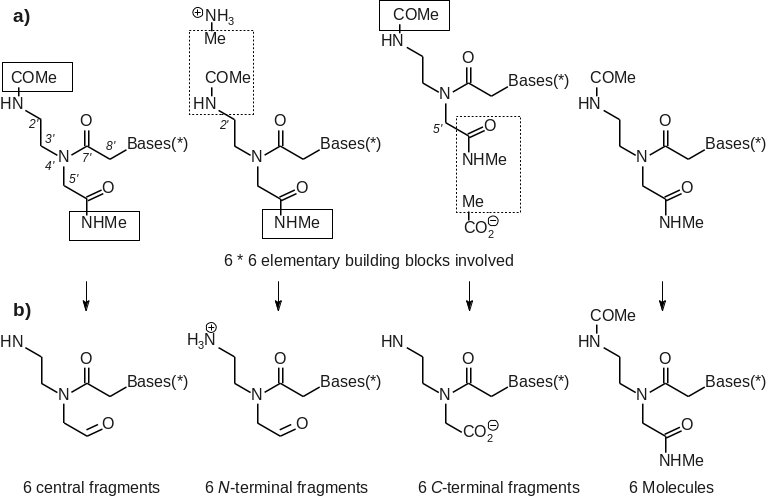 Figure 1
Figure 1
The statistics module of the R.E.D. IV program was used to characterize and minimize the impact of the charge constraints used during the charge fitting step. A RRMS value of 0.041 between the MEP calculated by quantum chemistry and that generated using the derived charge values was obtained for the charge fitting step. A highly similar RRMS value was also obtained in the absence of intra-molecular charge constraints, inter-molecular charge constraints and inter-molecular charge equivalencing. The relative small RRMS values as well as the minor difference of RRMS value between the charge fitting steps carried out with and without these charge constraints is one of the ways to demonstrate the accuracy of the fitting step performed in this work and the relative weak effect of the constraints used. Finally, rounding off errors of charge values were corrected at the fourth decimal point. RESP charges were validated by molecular dynamics simulations in condensed phase conditions. Structural features as well as melting temperatures for RNA-PNA heteroduplexes were found to be in agreement with experimental data. Wavelength scans from circular dichroism experiments of 12mer PNA-RNA duplexes showed A-helix signatures. Analysis of 50 ns NVT molecular dynamics simulations of 12mer PNA-RNA duplexes also produced A-like helical properties in agreement with experiments.
This R.E.DD.B. project provides a new FFTopDB for regular PNA with a highly homogeneous set of RESP charge values fully compatible with other molecular fragments belonging to the Amber FFTopDB for proteins and nucleic acids. A
script allowing loading the PNA force field libraries within the LEaP program as well as missing
force field parameters are also provided. The approach described here can be easily extended to other PNA structures such as γ-modified PNA, and will lead to the extension of the current FFTopDB.
[11]
[1] Egholm et al. J. Am. Chem. Soc. 1992, 114, 1895–1897. Egholm et al. Nature 1993, 365, 566–568.
[2] He et al. J. Am. Chem. Soc. 2008, 130, 13264–13273.
[3] Cornell et al. J. Am. Chem. Soc. 1995, 117, 5179–5197; Hornak et al. Proteins 2006, 65, 712–725.
[4] Cieplak et al. J. Comput. Chem. 1995, 16, 1357–1377.
[5] Dupradeau et al. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. Vanquelef et al. Nucl. Acids Res. 2011, 39, W511–W517.
[6] Brown et al. Science, 1994, 265, 777–780.
[7] Backbone and the nucleobase dihedral values for the PNA building blocks can be deduced from the corresponding optimized geometries provided to the PDB file format in this R.E.DD.B. project. All the building blocks present homogeneous optimized geometries except the guanine one, which displays a characteristic hydrogen bond between the N2 - O1' (Donor - Acceptor) atoms.
[8] Atoms involved in the RBRA procedure before MEP computation are defined as it follows: molecule number (1-36): total number of molecular orientations; three atom numbers separated by the pipe character: 1, 3, 5, 6, 7, 9, 11, 12, 13, 15, 17, 18, 19, 21, 23, 24, 25, 27, 29, 30, 31, 33, 35, 36: 4: 15 21 23 | 23 21 15 | 15 23 21 | 21 23 15; 2, 4, 8, 10, 14, 16, 20, 22, 26, 28, 32, 34: 2: 1 5 7 | 7 5 1. These pieces of information can also be obtained from the PDB files provided in this R.E.DD.B. project.
[9] Inter-molecular charge equivalencing between building blocks applied during the charge fitting stepare are defined as it follows: molecule numbers | atom numbers in the set of molecules: 1 7 13 19 25 31 | 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22; 2 8 14 20 26 32 | 5 6 7 8; 4 10 16 22 28 34 | 5 6 7; 3 9 15 21 27 33 | 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22; 5 11 17 23 29 35 | 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22. These pieces of information can also be obtained from the RESP input files provided in this R.E.DD.B. project.
[10] Bayly et al. J. Phys. Chem. 1993, 97, 10269–10280, and here.
[11] Crawford et al. J. Nucleic Acids 2011, 2011, 652702.





